Introduction
Chronic obstructive pulmonary disease (COPD) is the third leading cause of mortality, responsible for 3.23 million deaths per year worldwide [1]. Approximately 90% of COPD deaths among persons under 70 years of age occur in low or middle income countries [2]. COPD is characterized by persistent airflow limitation on spirometry and is caused by a complex interplay of environmental exposures with susceptible genetic profiles of individuals over time (i.e., gene-environment-time, GETomics) [3]. Typically, patients experience symptoms of COPD (e.g., cough, exertional dyspnea, or sputum production) in the fifth or sixth decades of life [1]. COPD is also punctuated by periods of acute worsening of symptoms and lung function, lasting several days to weeks at a time [1]. These episodes are called COPD exacerbations and in extreme cases can lead to emergency department visits, hospitalizations, and even mortality [1]. In high income countries, COPD exacerbations are the leading cause of hospitalization and rehospitalization [4]. Interestingly, although COPD exacerbations are usually triggered by an acute viral respiratory tract infection (and not bacterial infections), they are typically treated with antibiotics with or without systemic corticosteroids [5]. The exact mechanism(s) by which antibiotics improve health outcomes in COPD exacerbations are largely unknown. Interestingly, even in patients with stable disease (i.e., free of exacerbations for at least a month), use of antibiotics, especially macrolides, leads to better outcomes, reducing the frequency of COPD exacerbations by approximately 25% [6]. While the benefits of macrolides and other antibiotics in this setting may be related to their anti-inflammatory properties, it is possible that their antimicrobial effects may also be highly relevant. Traditionally, the airways below the vocal cords were thought to be sterile [7]; however, modern genomic technology has unveiled a rich and diverse microbial communities in the airways from the nose to the alveoli, which become perturbed in COPD, causing dysbiosis. In this review, we will review our current understanding of the airway microbiome (microbiota) in health and disease and discuss the impact of dysbiosis on morbidity and mortality of patients with COPD.
Methods
Using search terms ŌĆ£COPD,ŌĆØ ŌĆ£chronic obstructive pulmonary disease,ŌĆØ ŌĆ£microbiome,ŌĆØ and ŌĆ£bacteria,ŌĆØ PubMed was interrogated to January 1, 2023 to identify relevant papers on this topic. In addition, the reference lists of review papers and published systematic reviews were searched for relevant articles that may have been missed during the electronic search.
Definitions
For this review, we will use the Global initiative for chronic Obstructive Lung Disease (GOLD)ŌĆÖs definition of COPD, which is based on forced expiratory volume in 1 second/forced vital capacity (FEV1/FVC) ratio of less than 0.70 in the presence of chronic respiratory symptoms such as dyspnea, cough, or sputum production [8]. The microbiome is defined as a collection of genomes of all microorganisms in a specific anatomical location. Thus the airway microbiome denotes all microorganisms, dead or alive, in the lower respiratory tract [9]. Microbiota, on the other hand, refers to a collection of microorganisms that are alive and residing in a specific anatomical location [9]. The distinction between the two is subtle but important. As modern genomic techniques detect nucleic acids, which can be released by viable and nonviable microorganisms, in this review, we will use the term microbiome when referring to a collection of microorganisms in a specific location. Dysbiosis refers to an imbalance in the microbiome that is associated with a diseased state (Table 1).
Modern Molecular Techniques for Detection the Airway Microbiome
Traditionally, airway microbial flora were ascertained using culture methods of sputum or bronchoscopic samples (Table 2 for details). Typically, once the samples are collected from patients, these would be sent to a microbiology laboratory wherein the samples would be plated in a broth and/or agar-based media to encourage the growth of (pathogenic) bacteria. This method enabled not only the identification of potential pathogens but also antimicrobial susceptibility testing on that pathogen. Although this approach is considered the ŌĆ£gold standardŌĆØ for identifying airway microflora, there are many shortcomings to this approach that limit its application. For example, the culture methods bias the findings towards identification of bacteria that readily proliferate under laboratory conditions; they also provide qualitative rather than quantitative results and prevent ascertainment of bacteria and other microbial organisms that are fastidious and do not easily grow outside of the human airways. As most microorganisms in the airways do not grow in traditional bacterial media, the dogma in respiratory medicine has been that the airway below the vocal cords was ŌĆ£sterileŌĆØ in health [7]. Thus, cultivation of bacteria in sputum and/or bronchoscopic samples was considered pathogenic or a consequence of oral contamination.
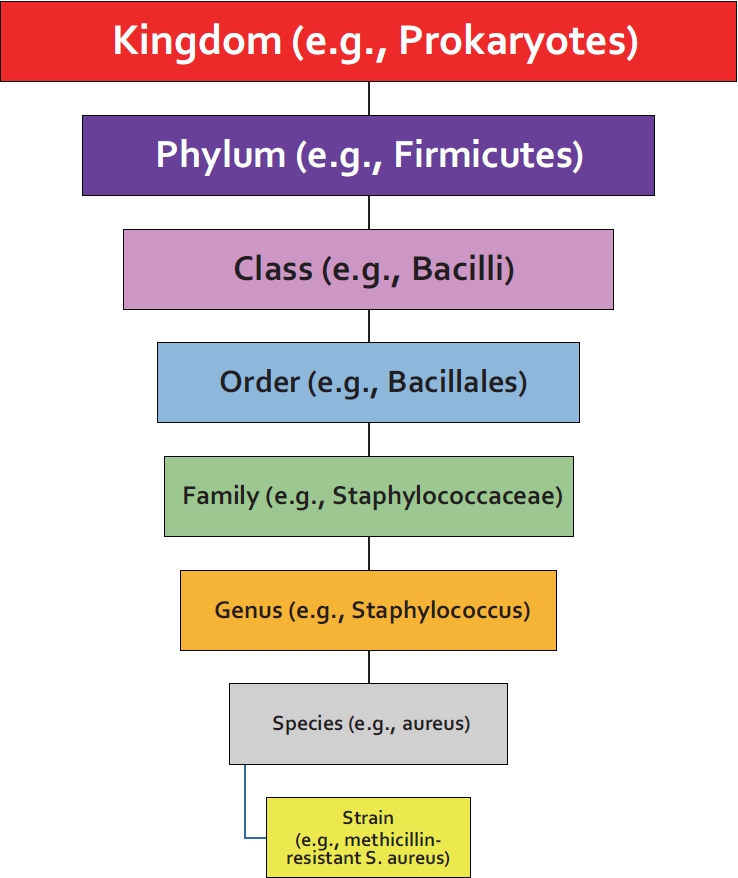
The advent of genomics technology such as polymerase chain reaction, DNA fingerprinting and next generation sequencing (NGS) has radically transformed the study of airway microbiome and revealed a rich and diverse microflora, though low in abundance, that were not previously recognized in health. All of these techniques rely on the fact that bacteria harbor a single gene that is highly conserved (i.e., 16S small subunit ribosomal RNA [rRNA]) [10]. The 16S rRNA codes for the 30S subunit in prokaryotic ribosomes and is composed of nine hypervariable regions (V1-V9), which are interspersed with conserved regions [10]. Because the V3 and V4 regions of the 16S rRNA demonstrate the greatest variability, sequencing these regions enables identification of most bacteria that are present in the sample of interest. Most investigators now use NGS (typically using an Illumina platform, San Diego, CA, USA) for high throughput sequencing of the 16S rRNA gene. This approach provides several million 250 base pair (bp) paired-end reads per flow cell at a relatively low cost. The main disadvantage of this approach is that the reads are relatively short and cannot be used to fully distinguish one type of bacteria from another [10]. Thus, this approach produces accurate results at a very high phylogenetic level (e.g., at the level of phylum) but are poor at discriminating communities of bacteria at a species or a strain level (see Figure 1 for explanation of the taxonomic hierarchy of bacteria). The introduction of long-read NGS technology (e.g., single molecule real time sequencing) may be able to overcome this limitation and provide accurate information on the diversity of bacteria to the species and strain level [10].
Fungal identification is performed similarly except that internal transcribed spacer (ITS) rather than 16S rRNA is used as the ŌĆ£barcodeŌĆØ for sequencing. ITSŌĆÖs are small spacer DNA, which are located in between the small subunit rRNA (18S) and the large subunit rRNA (28S) genes [11]. It should be noted; however, that the bioinformatics tool to annotate fungal elements in a sample is less robust and less developed than those used for bacterial identification.
Metagenomics, on the other hand, does not rely on ŌĆ£barcodeŌĆØ sequences [12]. Rather, it sequences all nucleic acids in a sample, regardless of whether they represent host or microbial DNA. As there is no bias introduced by barcode sequences, metagenomics produces the most robust and direct estimates of the microbial community composition and diversity within a specific sample [12]. Moreover, metagenomics enables identification of new organisms or new genes or proteins associated with a specific microbial communities that were previously missed by using more traditional methods. While the term metagenomics is reserved for the study of DNA, metatranscriptomics refers to sequencing of all RNA materials in a given sample [13]. As RNA is produced by living (micro) organisms, metatranscriptomics is used to determine which microbes are ŌĆ£activeŌĆØ and what they are producing; thus this approach enables a comprehensive evaluation of the microbiota. The major limitation of metagenomics and metatranscriptomics in the airways is the high cost of the procedure (e.g., >5,000 US dollar per sample to perform ŌĆ£shotgunŌĆØ metagenomics), the low biomass content of microorganisms relative to the human genomic materials in an airway sample, and the complex bioinformatics pipeline required to analyze the data [12,13].
In general, all of these genomic technologies share certain challenges. These include: (1) relatively high background noise resulting from potential genomic contamination of environmental microorganisms, high sequence stochastic noise, and contamination of organisms from the upper airway; (2) low microbe to host genetic content (described previously); and (3) technical errors including batch effects (i.e., noise related to systematic technical differences between experiments).
What Is the Normal Airway Microbiome?
Before embarking on what is abnormal, it is crucial to understand what a normal airway microbiome looks like. In general, the lower airway microbiome more closely resembles those of the oropharynx than those of the nasopharynx or other parts of the respiratory tract [14]. The most widely accepted postulate on the connection between the oropharyngeal microflora and the lower airway microbiota is microaspiration, complemented by direct dispersion of the microorganisms along contiguous mucosa from the upper to the lower airways. Although microaspiration occurs in everyone across the full age spectrum, most of the aspirated specimens are rapidly eliminated from the lower airway through a variety of different defense mechanisms including mucociliary clearance, coughing, and local immune responses (e.g., defensins and macrophages) [15]. However, with breakdown in the defense mechanisms (e.g., mucociliary escalator), microorganisms that would otherwise be expelled find residence in the lower respiratory tract (e.g., Pseudomonas species). In terms of absolute numbers, the oropharynx contains the greatest abundance of bacteria. Indeed, the oral microbiome is the second most diverse microbial community in the human body, next only to the gut microbiome [16]. The lower airway has very little burden of bacteria; its abundance is one million fold lower than that of the gut and 100-fold lower than that of the upper airway. In general, the abundance of bacteria progressively decreases as airways become progressively smaller [14]. However, there is an interesting ŌĆ£paradoxŌĆØ that should be noted. Although BAL fluid samples more distal airways than bronchial brushes, greater bacterial abundance is typically observed in BAL than in brush specimens. This is because by design bronchial brushings sample only approximately 1 cm2 of the airway tree. In contrast, BAL samples approximately 17,500 cm2 of the distal airways [14]. Interestingly, although the oropharynx contains many more microorganisms than the lower respiratory tract, there is more heterogeneity in the abundance of different microorganisms in the upper than in the lower airways [14]. In both upper and lower airways, the most abundant bacteria are in the Bacteroidetes, Proteobacteria, and Firmicutes phyla (Figure 2) [14,16,17]. Together, they constitute >90% of the observed species in the airway microbiome [14,16,18]. The oropharyngeal microbiome, however, contains higher levels of Firmicutes than in the lower airways [14,16,18]. At the genus level, Prevotella and Veillonella organisms constitute approximately 50% of the microbiome in the upper and lower airways [14,16,18].
In general, the airway microbiome in healthy persons is well-balanced [19]. As the airways become more diseased, the microbial balance becomes more disturbed. A commonly used metric to summarize the diversity in a particular site is alpha- (╬▒-diversity) and beta-diversity (╬▓-diversity) [9]. Alpha-diversity is defined as the mean diversity of microbial species in an ecological niche with respect to its richness (i.e., number of taxonomic groups) and evenness (i.e., distribution in the abundances across these taxonomic groups) [9]. With virtually all lung diseases and insults, ╬▒-diversity decreases relative to the healthy airway. ╬▓-Diversity, on the other hand, measures the number of different bacterial communities that are present in an ecological niche [9]. Thus, ╬▓-diversity enables comparison of diversity in bacterial communities between ecological niches (e.g., lower versus upper airway) and across different diseases (e.g., a comparison in lower airway microbiome between patients with COPD and healthy controls).
Airway Microbiome in COPD and Other Lung Diseases
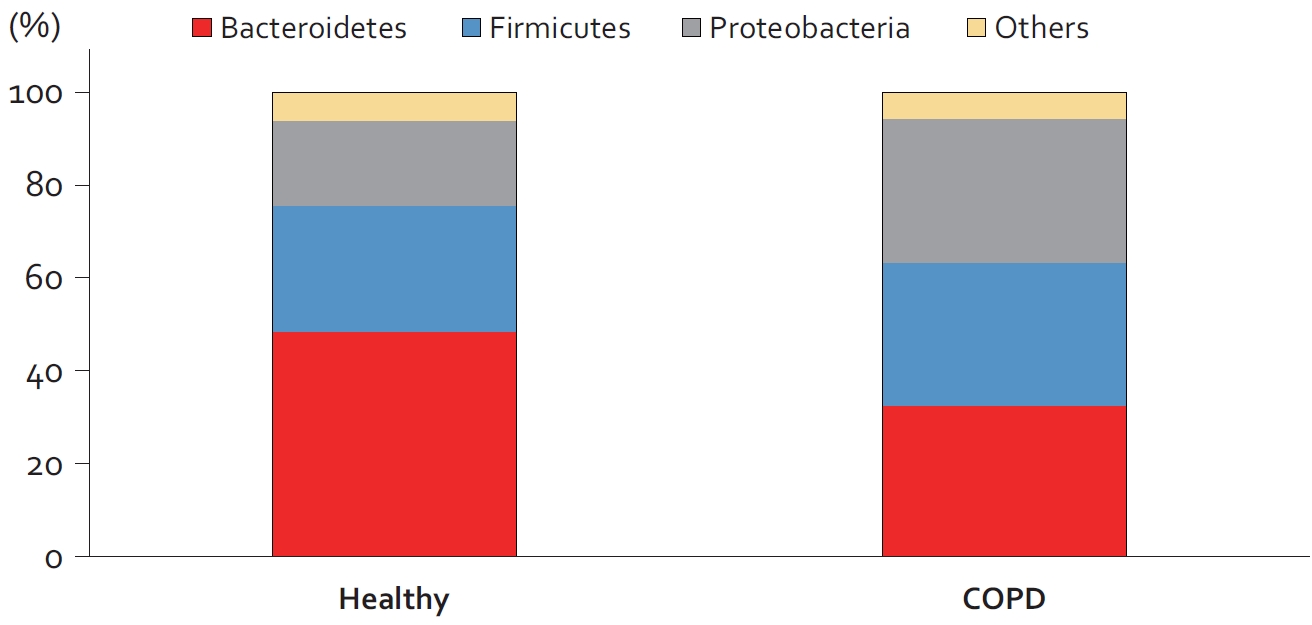
Although the total abundance of microorganisms in the upper and lower airways is similar between patients with COPD and healthy controls, there is dysbiosis in the lower airways of patients with COPD [20]. As compared with the healthy airway, the COPD airway is less diverse (i.e., decreased ╬▒-diversity driven largely a reduction in the number of observed species in the COPD airway) and contains more bacteria in the phyla Proteobacteria, Firmicutes, and Actinobacteria and less members in the Bacteroidetes phylum [20] (Figure 3). At the genus level, the COPD airways contain more bacteria in the Hemophilus genus. Interestingly, whereas the genus Moroxella is rarely observed in the lower airways of healthy control subjects, these bacterial members are present in approximately 2% of airways of patients with COPD [20]. Although there is some heterogeneity of data across studies, the totality of data suggest that the relative abundances of Hemophilus, Moraxella, and Pseudomonas are increased in COPD and that of Prevotella becomes depleted in COPD [18].
Not only does ╬▒-diversity (and in particular richness) decrease with increasing airflow limitation, it also declines with increasing burden of emphysema that leads to a reduction in total alveolar surface area [21]. In GOLD 4 disease (i.e., FEV1<30% of predicted), there is expansion of bacteria in the Proteobacteria phylum [21].
Because bronchoscopic and lung surgery methods of tissue specimen acquisition are invasive and fraught with potential harm to patients, spontaneous or induced sputum collection has been used to evaluate the airway microbiome in patients with COPD. The main limitations of this approach are the challenges of obtaining good quality samples, possible contamination by upper airway flora and low diagnostic yield. Nevertheless, a few studies using sputum samples have provided great insights on the role of airway microbiome in the pathogenesis of COPD.
In the largest study of its kind to date, Yan et al. [22] performed metagenomics profiling of induced sputum samples in 72 patients with COPD and 18 healthy control subjects. They also performed metabolomic and transcriptomic profiling of sputum specimens in a vast majority of these individuals. Using the sputum metagenome, the investigators found that modules for bacterial transport system and amino acid biosynthesis were enriched in COPD samples; whereas modules for energy metabolism and breakdown of amino acids were depleted in COPD samples. Thus, the sputum metagenome suggested that the COPD airway promotes amino acid anabolism (rather than catabolism) relative to the healthy airways [22]. These data were largely confirmed on sputum metabolomics, which showed that COPD samples were enriched for metabolites of amino acids, carbohydrates, and steroids. In contrast, COPD samples were depleted for metabolites of phosphatidylcholines [22]. These investigators also interrogated pathways relating to neutrophilic inflammation in airways of COPD patients and showed that altered tryptophan metabolism, resulting in depletion of indole-3-acetic acid (IAA), may promote airway neutrophilia and epithelial cell apoptosis. Interestingly, they showed using a murine model that inoculation of mice with Lactobacillus species of bacteria increased IAA levels in the airway, which protected these mice against tissue injury, apoptosis, neutrophilic inflammation and most importantly emphysema and lung function decline when these animals were exposed to long-term cigarette smoke or lipopolysaccharide (LPS) [22]. Together, these data suggest that the airway microbiome plays a significant role in the pathogenesis of neutrophilic inflammation of COPD.
Dicker et al. [19] performed 16S sequencing in spontaneous sputum of 253 clinically stable patients with COPD and followed these patients for a median of 4 years. They found that sputum demonstrating lower diversity (as measured by ╬▒-diversity) and containing a predominance of bacterial members from the Proteobacteria phylum had lower lung function and most importantly were at increased risk of COPD exacerbations. They also showed that the Proteobacteria predominant sputum demonstrated neutrophil activation in the airways [19].
These data are consistent with older data showing that culture positivity for potential pathogenic bacteria such as Hemophilus influenzae, Streptococcus pneumoniae, and Pseudomonas aeruginosa was associated with increased risk of exacerbations [23]. In the largest study of its kind, Rosell et al. [23] performed bronchoscopy in patients with COPD and sent bronchoscopic samples for traditional bacterial cultures. They found that approximately 25% of ŌĆ£stableŌĆØ patients were colonized with a potential pathogenic bacteria at a concentration of 100 colony-forming units/mL or greater. The most common organisms were H. influenzae and S. pneumoniae. The rate of colonization was higher in those who exacerbated (approximately 50%) with a predominance of H. influenza and P. aeruginosa [23]. These data are also consistent with those of Alotaibi et al. [24], who applied the Randox (Crumlin, UK) cystic fibrosis respiratory pathogen panel that probes for the nucleic acid of 21 different bacteria, seven viruses, and four fungi on BAL cell pellets obtained from patients with stable COPD. Using a modern nucleic acid detection method (rather than relying on traditional culture methods), they found that nearly 50% of the samples contained one or more of these potential pathogens and the presence of these potential pathogens was associated with significant neutrophilia in the BAL fluid and at the patient level, worse health status as measured on the St. GeorgeŌĆÖs Respiratory Questionnaire [24]. Together, the studies by Dicker et al. [19], Rosell et al. [23], and Alotaibi et al. [24] suggest that the airway microbiome plays a significant role in the pathogenesis and risk of COPD exacerbations.
Another important observation by Dicker et al. [19] was that the airway dysbiosis is associated with increased mortality in patients with COPD. They showed that Proteobacteria predominance in the sputum of patients with COPD was associated with increased mortality compared with patients who demonstrated a predominance of Firmicutes. The lowest risk of mortality was observed in patients who had ŌĆ£balancedŌĆØ microbiome profiles in their sputum [19].
Leitao Filho et al. [25] extended these data from Dicker et al. [19] by performing 16S sequencing on spontaneous sputum specimens collected at the time of severe COPD exacerbations in hospitalized COPD patients. They showed in 102 patients that patients who had lower ╬▒-diversity in sputum had 3-fold increased risk in 1-year mortality compared with patients who had higher ╬▒-diversity. Importantly, they demonstrated that mortality was significantly related to depletion of bacteria in the Veillonella genus and increased abundance of Staphylococcus bacteria. Indeed, patients whose sputum was negative for Veillonella and positive for Staphylococcus were 85 times as likely to experience mortality over 1 year as patients who were positive in their sputum for Veillonella and negative for Staphylococcus [25].
The Impact of Therapeutics on the Airway Microbiome in COPD
Azithromycin (at a low-dose, 250 mg/day) is a commonly used macrolide antibiotic to prevent exacerbations in patients with COPD, who frequently exacerbate despite maximal inhaled therapies [8]. To determine whether low-dose azithromycin therapy changes the airway microbiome of patients with COPD, Segal et al. [26] treated stable COPD patients with a 8-week course of azithromycin (250 mg/day) or placebo and performed bronchoscopy at baseline and at the end of the 8-week treatment period. During bronchoscopy, BAL was performed in either the right middle lobe or the lingular segment and 16S sequencing was performed on the BAL fluid. The investigators found that 8 weeks of low-dose azithromycin therapy led to significant changes in the ╬▓- but not ╬▒-diversity of the BAL samples (compared with placebo). Azithromycin therapy also significantly reduced the expression of bacteria in Tissierellaceae, Cytophaga, Flectobacillus, Neisseria, Ralstonia, and Rhodospirillaceae and increased the representation of membership in Acidimicrobiales, Bifidobacterium, and Bradyrhizobiaceae. Interestingly, the BAL fluid of patients treated with low-dose azithromycin demonstrated increased abundance of metabolites, benzoic acid, glycolic acid, indole-3-acetate, and linoleic acid [26]. Treatment with azithromycin also resulted in lower concentrations of inflammatory cytokines such as tumor necrosis factor ╬▒ (TNF-╬▒), interleukin 12 (IL-12) p40, IL-13, and chemokine (CXC motif) ligand 1 (CXCL1) in the BAL fluid of patients with COPD. Ex vivo, treatment of alveolar macrophages with glycolic acid or idole-3-acetate directly suppressed the production of TNF-╬▒, IL-12 p40, IL-13, and CXCL1 in response to LPS [26]. Together, these data suggest that azithromycin directly modifies the community composition of bacteria in COPD airways and suppresses inflammation by increasing microbial metabolites such as indole-3-acetate.
Inhaled corticosteroids (ICSs) are the most widely used anti-inflammatory therapies in patients with COPD [27]. Although they improve health status and FEV1 and significantly reduce the risk of exacerbations, their use has also been associated with increased risk of pneumonia in certain patients [28]. In a large cross-sectional study, Ramsheh et al. [20] performed bronchoscopies in 360 patients with COPD and showed that the use of ICS did not modify ╬▒- or ╬▓-diversity in bronchial brush specimens. However, the mean abundance of bacteria in Proteobacteria phylum was significantly higher and that of bacteria in Bacteroidetes phylum was significantly lower in patients who used ICS compared with those who did not. At the genus level, patients who were not treated with ICS had higher abundance of both Prevotella and Veillonella than those who were treated with ICS [20].
Leitao Filho et al. [18] extended these findings by performing a randomized controlled trial to determine the effect of ICS on the airway microbiome of patients with COPD (Study to Investigate the Differential Effects of Inhaled Symbicort and Advair on Lung Microbiota [DISARM] Trial) [29]. In this trial, patients first underwent bronchoscopy when they were free of any ICS for at least 4 weeks prior to the procedure. A week later, they were randomized to a ICS/long-acting beta-2 agonist (ICS/LABA) arm or a LABA only group. After 3 months of treatment, they underwent a second bronchoscopy. Bronchial brushes were obtained from the same subsegment in both the first and second bronchoscopies. Bronchial brush specimens from patients treated with ICS demonstrated lower ╬▒-diversity (especially a large reduction in microbial richness) over 3 months compared with patients treated with LABA only. They also found that ╬▓-diversity also changed with ICS/LABA therapy, especially that containing fluticasone. The greatest reduction in relative abundance was noted for bacteria in the order of Pasteurellaies, genus Hemophilus, and genus Alloprevotella [18]. Together, these studies indicate that ICS therapy reduces ╬▒-diversity and modifies the community composition of the airway microbiome in patients with COPD. Whether these observations explain the increased risk of pneumonia related to ICS are unknown.
Conclusions
Contrary to traditional teachings, the lower airway is not sterile and contains a rich and diverse microflora. However, relative to other organs such as the skin and the gut, the overall abundance of bacteria in the lower airways is low and can only be reliably detected using modern molecular techniques (e.g., NGS). Members in three phlyla (i.e., Bacteroidetes, Firmicutes, and Proteobacteria) dominate the lower airways. In patients with COPD, there is a reduction in ╬▒-diversity and increased representation of bacteria from Firmicutes and especially Proteobacteria. Increased abundance of Proteobacteria members and relative reduction in Bacteroidetes bacteria have been associated with increased mortality and exacerbations. ICS therapy may reduce ╬▒-diversity and shift the community composition of bacteria in the lower airways of COPD patients. Azithromycin also modifies the airway microbiome and increases microbial metabolites such as IAA, which may suppress airway inflammation. Novel therapeutics are urgently needed to specifically target the airway microbiome and improve health outcomes of patients with COPD.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Print
Print Download Citation
Download Citation