The Role of Innate and Adaptive Immune Cells in the Immunopathogenesis of Chronic Obstructive Pulmonary Disease
Article information
Abstract
Chronic obstructive pulmonary disease (COPD) is a chronic and progressive inflammatory disease of the airways and lungs that results in limitations of continuous airflow and is caused by exposure to noxious gasses and particles. A major cause of morbidity and mortality in adults, COPD is a complex disease pathologically mediated by many inflammatory pathways. Macrophages, neutrophils, dendritic cells, and CD8+ T-lymphocytes are the key inflammatory cells involved in COPD. Recently, the non-coding small RNA, micro-RNA, have also been intensively investigated and evidence suggest that it plays a role in the pathogenesis of COPD. Here, we discuss the accumulated evidence that has since revealed the role of each inflammatory cell and their involvement in the immunopathogenesis of COPD. Mechanisms of steroid resistance in COPD will also be briefly discussed.
Introduction
In 2020, chronic obstructive pulmonary disease (COPD) will be the third leading cause of death worldwide (from sixth in 1990) and fifth leading cause of years lost through early mortality or handicap (disability-adjusted life years) from previously 12th in 19901. However, recent studies suggest COPD is already the third most common cause of death, worldwide2. For the population without COPD over the age of 40, the risk of developing COPD within the next 40 years was 12.7% for men and 8.3% for women3. In patients with very severe COPD, 26% died after 1 year of follow-up, whereas 2.8% died among the non-COPD subjects3.
COPD, a common and preventable disease, is characterized by persistent airflow limitation that is usually progressive and associated with an enhanced chronic inflammatory response in the airways and the lung to noxious particles or gasses4. The Global Initiative for Chronic Obstructive Lung Disease (GOLD) defines airflow obstruction as spirometry where the ratio of forced expiratory volume in the first second to forced vital capacity after bronchodilation is less than 0.705.
Cigarette Smoke Exposure as a Model to Study COPD
Cigarette smoking (CS) is an established risk factor for COPD67, and study suggests that CS exposure could have a suppressive effect on host innate immunity including structural and functional changes in the respiratory ciliary epithelium, and immune cells such as alveolar macrophages (AMs), neutrophils, and lymphocytes8. Moreover, CS could also cause defect in the generation of adaptive immunity in the lung9. CS exposure is the most appropriate model to study the pathogenesis of emphysema in mice10. Following cigarette exposure for 26, 52, and 65 weeks, structural changes were observed and accompanied by altered lung function at 26 and 52 weeks11. After 13 weeks of CS exposure-free period, most biochemical, histopathological, and morphometrical alterations were restored, while emphysema was observed to persist in 18% of mice exposed to CS at 65 weeks. These findings suggest that the cigarette exposure induced emphysematous changes in the lungs, accompanied by altered lung function and inflammatory cell infiltration. There are data demonstrating that smoking is associated with up- or down-regulation of many genes in the airway epithelium and, interestingly, ex-smokers continue to have persistent up- or down-regulation of many genes, despite smoking cessation12.
Early Changes in Airway Epithelium in the Pathogenesis of COPD
Most of the normal human airway is lined by a pseudostratified epithelium of ciliated cells, secretory cells and 6%-30% basal cells (BCs)13. In COPD, the remodeling of the airway epithelium, such as squamous metaplasia and mucous hyperplasia that occur during injury, may considerably disturb the innate immune functions of the airway epithelium14. Although inflammatory reaction by immune processes plays a significant role in the pathogenesis of COPD, the earliest abnormalities in the COPD lung caused by smoking are hyperplasia of airway BC, the stem/progenitor cells of the ciliated and secretory cells that are central to pulmonary host defense15. Apart from BC hyperplasia, smoking induces a number of COPD-relevant airway epithelial remodeling phenotypes that are likely initiated in the BC population, including mucous cell hyperplasia, squamous cell metaplasia, epithelial-mesenchymal transition, altered ciliated and non-mucous secretory cell differentiation, and suppression of junctional barrier integrity16.
Inflammatory Cells in COPD
A smoking-induced inflammatory reactions in the airways and lung parenchyma have long been accepted to be the major cause of COPD in smokers17. Cigarette smoke activates innate immune cells by triggering pattern recognition receptors to release "danger signal" that act as ligands to Toll-like receptors, triggering the production of cytokines and inducing innate inflammation18.
Impaired immune function contributes to the development of COPD and disease progression is further exacerbated by infections due to impaired immune responses19. Severity and course of acute exacerbations of COPD reflects the capacity of the adaptive immune system in modulating the innate response to pathogen ("the Goldilocks hypothesis")20. Basically, this hypothesis states that there is no such thing as bad inflammation; it all depends on how and when21. Goldilocks hypothesis is the state when the innate and adaptive immune response successfully eliminates the infection, and the response is mild and transient, not too much and not too little20. The immune inflammatory changes associated with COPD are linked to a tissue-repair and -remodeling process that increases mucus production and causes emphysematous destruction of the gas-exchanging surface of the lung22.
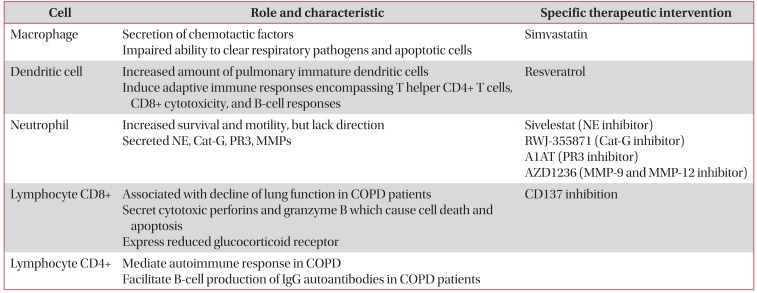
The inflamed airways of COPD patients contain several inflammatory cells including neutrophils, macrophages, T lymphocytes, and dendritic cells (DCs)23 (Table 1). Recent evidence also found increased population of goblet cells in COPD patient24, which cause the mucous overproduction and hypersecretion25. It seems likely that, only when all inflammatory cell types (i.e., CD4+, CD8+, neutrophils, and macrophages) are present in the lung, the airways remodeling and parenchymal destruction characteristic of COPD will ensue17. These cells release the reactive oxygen species, chemokines (e.g., interleukin [IL]-8), cytokines (e.g., tumor necrosis factor alpha [TNF-α]) and proteases (e.g., neutrophil elastase [NE] and matrix metalloproteinase [MMP]) that are instrumental in producing a chronic inflammatory state26. In the following sections, we will discuss each inflammatory cell that participates in the immunopathogenesis of COPD.

The role of innate and adaptive immune cells in the pathogenesis of COPD
Macrophage
AMs have been identified as one of the major cell types that plays a key role in orchestrating the inflammatory events associated with the pathophysiology of COPD27. The number of AM is markedly increased in the lungs of patients with COPD as a result of increased recruitment, proliferation and survival28. One of the major functions of macrophages is the secretion of chemotactic factors and this function is markedly increased on exposure to CS27. However, it is also found that macrophage in COPD has impaired ability to clear respiratory pathogens and apoptotic cells29. The reduced phagocytic ability of macrophage may drive the persistence of inflammation in COPD30.
Genome-wide analysis has underscored the heterogeneity and plasticity of macrophage phenotype which lead to the introduction of the term M1 (classical activation) and M2 (alternative activation)31. M1 phenotype macrophages express various pro-inflammatory mediators including TNF-α, IL-1, IL-6, reactive nitrogen and oxygen intermediates, which have a strong microbicidal and tumoricidal activity; while M2 phenotype is involved in tissue remodeling and characterized with anti-inflammatory properties32. It is said that the surrounding pulmonary environment in COPD may generate a specific phenotype that is permanently pro-inflammatory (M1)30. Among COPD patients, M2 macrophages were reduced in current smokers compared to ex-smokers which indicate that smoking cessation in COPD is associated with macrophage polarization towards an anti-inflammatory phenotype33. However another study suggests, rather than up-regulating the M1 polarization program as expected, CS induces in AM of COPD smokers the opposite phenotype, characterized by a substantial down-regulation of the M1-related genes34. Obviously, these conflicting results indicate that smoking induced a complex suppression of immune response in the lung, including deactivation of AM inflammatory and host defense function, and development of tissue remodeling.
The source of macrophage activation is still under intense investigation. Macrophage could be induced by TNF-α35 and IL-836. In one study by Bozinovski et al.37, activation of innate cellular sources of IL-17A is an essential mediator of macrophage accumulation in CS-exposed lungs. Targeting nonconventional T cell sources of IL-17A may offer an alternative strategy to reduce pathogenic macrophages in COPD. However, it has also been found that IL-17A contributes to normal lung homeostasis and does not mediate CS-induced loss of lung structure and pulmonary function38.
'Macrophage-targeted therapy' has been studied in many centers. It has been reported that simvastatin, member of statin which has immunomodulatory properties, reversed the IL-17A/IL-10 imbalance in the airways and reduced sputum macrophage but not neutrophil counts in patients with COPD39. Generally, simvastatin could provide substantial benefits in patients with COPD due to (1) reduction of cytokine secretion (TNF-α, IL-6, and IL-8) and neutrophil infiltration into the lung; (2) attenuation of the fibrotic activity in the lung leading to small airways fibrosis and irreversible airflow limitation; (3) antioxidant and anti-inflammatory effects on skeletal muscle; (4) reduced inflammatory response to pulmonary infection; and (5) inhibition of the epithelialmesenchymal transition, a precursor event to lung cancer40. In one in vivo study, pretreatment with simvastatin prior to and continued throughout smoke exposure reduced the influx of macrophages into the lung and airways41. Molecular mechanism of simvastatin in attenuating CS-induced emphysematous abnormality in COPD has been described by Kim et al.42. In their in vitro study, simvastatin reversed CS-induced MMP-9 expressions in AM. However, there are clinical trials that suggest simvastatin did not affect exacerbation rates or the time to a first exacerbation43, and did not reduce circulating inflammatory markers in patients with COPD44. Controversies about the use of statin in COPD still remain. Recently, positive result came from study conducted by Ingebrigtsen et al.4546, who found that statin was associated with reduced odds of exacerbations in COPD patient, in particular those who have coexisting cardiovascular disease. This finding was consistent with other study, involving 1,584 patients, that confirm the efficacy of statin in the reduction of COPD exacerbation47.
Dendritic Cells
DCs in the lung have an essential role in defense mechanism based on their anatomical location that creates a functional cellular interface between the external environment and the internal lung microenvironment48. DCs are in charge of capturing antigens in peripheral tissues, transporting them to lymph nodes and presenting them on major histocompatibility complex molecules to T lymphocytes49. DCs are professional antigen presenting cells linking innate and adaptive immune responses50. Cigarette smoke promotes DCs accumulation 51, and induces the release of chemokines from DCs that play a role in the pathogenesis of COPD52. It has been reported that COPD patients have an increased amount of pulmonary immature DCs. Using CD83+ and DC-LAMP(+) cells as mature DC marker, Tsoumakidou et al.53, found that mature DCs were decreased in non-COPD subject when they actively smoke cigarette and increased when they ceased smoking. They also found that COPD patients have a low mature DCs compared to never-smokers and ex-smokers non-COPD subject. Activated DCs induce adaptive immune responses encompassing T helper (Th1 and Th17) CD4+ T cells, CD8+ cytotoxicity, and B cell responses, which lead to the development of lymphoid follicles on chronic inflammation54. Moreover, remarkable up-regulation of CD80 and CD86 and secretion of cytokines interferon (IFN)-α were observed in DCs from COPD patients55. One of the drugs that has recently been found to inhibit dysfunction of DC is resveratrol55. Results of that study suggest that small doses of resveratrol are sufficient to inhibit phenotypic and functional TNF-α effect on DC56.
Neutrophil
Neutrophils are also aberrant with increased survival and motility, but lack of direction could lead to more widespread destruction of host matrix during migration30. Neutrophil secreted NE and the proteolytic activity of NE not only destroys pathogens but also degrades host matrix tissues by generating a localized protease-antiprotease imbalance in COPD57. Although NE was identified as a therapeutic target for COPD more than 30 years ago, only Sivelestat (ONO-5046), an NE inhibitor from Ono Pharmaceutical, has been approved for clinical use in Japan, but not in United States58.
There are two similar neutrophil serine proteases that coexist with NE in COPD: cathepsin G (Cat-G); and proteinase 3 (PR3)59. Cat-G is a neutral proteinase originating from human neutrophils that displays a unique dual specificity (trypsinand chymotrypsin-like); thus, its enzymatic activity is difficult to control60. Anti-inflammatory pharmacology of RWJ-355871, an inhibitor of Cat-G, has been tested in animal models of inflammation that represent COPD, and showed promising result in terms of reduction of smoke-induced neutrophilia61. Meanwhile, PR3 is a multifunctional serine proteinase mainly located in the azurophilic granules and on the cell surface of neutrophils and able to degrade many critical components of the extracellular matrix, including elastin, type IV collagen, fibronectin, and laminin62. In one report, PR3 is inhibited by the antiprotease α1-antitrypsin (A1AT)63.
MMPs have an NH2 terminal pro-domain, an active site zinc atom, and a COOH terminal hemopexin domain that regulates the binding of the enzymes to their substrates64. In cigarette smoke-induced animal models of emphysema, MMP-12 appears to play a consistent and important role, whereas the data for other MMPs are controversial65. Other studies suggest MMP-9 is associated with acute exacerbations of COPD66, and airway remodeling67. A phase IIA study has been conducted to evaluate the effects of AZD1236, a selective MMP-9 and MMP-12 inhibitor, in modifying differential cell count and in reducing TNF-α level in induced sputum68. Although AZD1236 was generally well tolerated over 6 weeks in patients with moderate-to-severe COPD, no clinical efficacy of AZD1236 was demonstrated in the study.
Recently, Taylan et al.69 reported that neutrophil-lymphocyte ratio (NLR) was altered in patients with COPD and could be used for identifying the severity of inflammation and recognition of acute exacerbation, similar to C-reactive protein and erythrocyte sedimentation rate. For an NLR cutoff of 3.29, sensitivity for detecting exacerbation of COPD was 80.8% and specificity was 77.7%69. Moreover, NLR values of the stable COPD patients were significantly higher than those of the healthy controls (p<0.001)70.
Lymphocyte CD8+
Decline in lung function in COPD patients has been correlated to the number of CD8+ T cells present in the lung as well as to a decline in the ratio of CD4+/CD8+ T cells71. Regardless of smoking habits, CD8+ T-cell activation was found in COPD, supporting the concept that this T-cell subset may play a role in the pathogenesis of COPD72. Smokers with COPD had a decreased ratio CD4+/CD8+ in the paratracheal lymph node compared to smokers without COPD73. Moreover, CD8+ T cells were found in both bronchial epithelium and airway lumen of COPD patients74. Lung CD8+ T cells might contribute to progression of COPD indirectly via IFN-γ production or directly via cytolysis, and in one in vitro study, stimulation of lung CD8+ T cells with IL-18 plus IL-12 markedly increased production of IFN-γ and TNF-α, whereas IL-15 stimulation induced increased intracellular perforin expression75. Cytotoxic perforins and granzyme B, produced by CD8+ T cells, can cause cell death and apoptosis which are feature of emphysema pathology76. Other CD8+ T-cell product, IFN-inducible protein-10, induces production of macrophage elastase (MMP-12) that degrades elastin, both causing lung destruction directly and generating elastin fragments that serve as monocyte chemokines augmenting macrophage-mediated lung destruction77. In vitro CD8+/CD28-cells were increased in both current- and ex-smoker COPD groups; under CS exposure, these cells expressed significantly more interferon IFN-γ, granzyme, and perforin compared with CS-exposed CD8+/CD28+ T cells78. Moreover, CD8+/CD28- cells have reduced glucocorticoid receptor which contributes to steroid resistance in COPD79. Recent evidence suggest that targeting CD137 expression in CD8+/CD28- lymphocyte T cells was associated with down-regulation of IFN-γ, TNF-α, and granzyme B80.
Lymphocyte CD4+
CD4+ T cells are important in amplifying inflammatory responses by other immune effector cells by means of promoting the long-term survival of CD8+ T cells and activating antibody-elaborating B cells81. Autoimmune responses mediated by CD4+ T cells may contribute to the development of COPD82. CD4+ T cells might facilitate B cell production of IgG autoantibodies in COPD patients, because ~70% of COPD patients had circulating IgG autoantibodies against epithelial cells compared to 10% among non-smoking controls and 13% of non-COPD cigarette smokers83. Increased CD4+/CD28-T cells in COPD indicates that chronic antigen exposure, e.g., through contents of smoke, leads to loss of CD28 and upregulation of natural killer cell receptors expression on T cells in susceptible patients84.
MicroRNA in COPD
MicroRNAs (miRNAs) are small non-coding RNA molecules that negatively regulate gene expression at the post-transcriptional level8586. Altered miRNA expression profiles may be associated with pathological processes within the lung and lead to the development of various pulmonary diseases, including inflammatory lung diseases87.
Among well studied miRNA, miR-15b, miR-223, and miR-1274a were the most important miRNAs in patients with COPD88 to both emphysematous and fibrotic areas and was differentially expressed according to the GOLD classification of COPD. miR-15b was increased in COPD samples compared with smokers without obstruction and localized to both emphysematous and fibrotic areas and was differentially expressed according to the GOLD classification of COPD. Meanwhile, miR-223 and miR-1274a were the most affected miRNAs in subjects with COPD compared with smokers without obstruction. miRNA-34c is associated with emphysema severity89. Healthy subject showed higher expression of let-7c and miR-125b compared with COPD subject90. Target gene of let-7c is tumor necrotizing factor receptor II (TNFR-II), and the study confirmed that the concentration of TNFR-II was inversely correlated with the sputum levels of let-7c.
Mechanism of Steroid Resistance in COPD
Resistance to the anti-inflammatory effects of corticosteroids is common in COPD91. Regular treatment with high doses of inhaled glucocorticoids does not significantly change the number of inflammatory cells in bronchial biopsies from patients with moderate COPD92. Several molecular mechanisms of glucocorticoid resistance have now been identified, including activation of mitogen-activated protein kinase pathways by certain cytokines, excessive activation of the transcription factor activator protein 1, reduced histone deacetylase-2 (HDAC2) expression, raised macrophage migration inhibitory factor, and increased P-glycoprotein-mediated drug efflux93. One of the novel strategies for overcoming steroid resistance is to increase HDAC2 expression by theophylline or phosphoinositide 3-kinase δ inhibitors94.
Several studies indicate that increased expression of IFN-γ has been correlated with macrophage-mediated steroid-resistant phenotype in chronic inflammatory disease9596. IFN-γ is a cytokine produced by Th1 cell and may play an important role in inflammation in individuals with COPD97, because it has been demonstrated that COPD patients have increased level of IFN-γ signaling in their lung98. Moreover, it has been shown that inhibition of IFN-γ signaling is a potentially novel strategy to sensitize macrophage to steroid in COPD99. Despite these findings, conflicting evidence also suggest that IFN-γ reversed steroid resistant in COPD through the restoration of dexamethasone-induced glucocorticoids-receptor α nuclear translocation in T cell100. Taken together, targeting IFN-γ as a mean to sensitize COPD patients to steroid need further investigations because of the wide variation of immune cells response to IFN-γ-targeted approach.
Lymphocyte has also been associated with steroid-resistant in COPD. One report suggests that T lymphocytes from the airways COPD patients display corticosteroid insensitivity via increased IFN-γ production101. Another work found that there is subset of lymphocyte population, CD8+/CD28- lymphocyte cells, that is resistant to steroid and this subset of population was increased in COPD79. Other mechanisms of lymphocyte-associated steroid resistance in COPD are reduced population of Treg cells (through reduced expression of IL-19 and vitamin D3) and increased population of IL17-producing Th1794.
Conclusion
COPD is a complex inflammatory disease of lung and exposure to CS is the most appropriate model to study the pathophysiology of COPD. The abnormalities in the earliest phase of smoking COPD are hyperplasia of airway BCs, the stem/progenitor cells of the ciliated and secretory cells that are central to pulmonary host defense. The airways of COPD patients have inflammatory cells including neutrophils, macrophages, T lymphocytes, and DC. Despite the inflammation characteristic of COPD, treatment with steroid does not significantly change the number of inflammatory cells due to several resistance mechanisms. Further studies are required to verify the effect of biological intervention on the inflammatory cells and find the ways to overcome the steroid resistance in COPD.
Notes
Conflicts of Interest: No potential conflict of interest relevant to this article was reported.