Introduction
Idiopathic nonspecific interstitial pneumonia (NSIP) is one of the varieties of idiopathic interstitial pneumonias (IIP). NSIP used to be considered as not an independent disease entity but provisional diagnosis. However, now, it is recognized as a distinct entity with clinical features that differentiate it from other interstitial pneumonias1,2. Diagnosis of idiopathic NSIP can be done via multidisciplinary approach in which the clinical, radiologic and pathologic findings are discussed together and other causes are excluded.
History and Epidemiology
1. History
NSIP began to gain attention in 1994 when Katzenstein and Fiorelli3 described histologic findings that did not fit the traditional Liebow classification of interstitial pneumonia. These pathology showed temporally uniform inflammation and fibrosis which distinguished them from other interstitial pneumonias, such as usual interstitial pneumonia (UIP). However, authors at the time concluded that it would be unreasonable to regard NSIP as an independent disease entity. In subsequent studies, the possibility was raised that NSIP might be independent disease entity although the histologic findings of NSIP were associated with a variety of causes including connective tissue disease (CTD)2. And, at the 2002 American Thoracic Society (ATS)/European Respiratory Society (ERS) international consensus classification, NSIP was classified as ŌĆ£provisional typeŌĆØ interstitial pneumonia4. In 2008, Travis et al.2 reported that idiopathic NSIP is a distinct clinical entity that is distinguishable from other interstitial pneumonias. They also reported that NSIP occurred mostly in non-smoking middle aged women, and its prognosis is good. In the recent revised ATS/ERS international consensus classification, idiopathic NSIP was classified as a distinct entity among the IIPs1.
2. Epidemiology
The incidence and prevalence of NSIP are not clearly known. However, according to several retrospective cohort studies, the prevalence has been estimated at 1 to 9 per 100,000 people5, and the incidence has been estimated at around 3 per one million people6. According to a nationwide survey conducted by the Korean Academy and Tuberculosis and Respiratory Diseases in 2008, NSIP was the second common IIP next to idiopathic pulmonary fibrosis (IPF), accounting for 11.9% of 2,186 patients with IIP7. In a recent cohort of a university hospital in Denmark, 431 cases of interstitial lung diseases were analyzed from 2003 to 2009. NSIP accounted for 7% of them. And it was the fourth most common interstitial lung disease following IPF, CTD interstitial pneumonia, and hypersensitivity pneumonitis (HP)6. NSIP is more common in females and non-smokers than their counterparts, and occurs at lower ages than IPF2,7.
Clinical Features and Diagnosis
1. Clinical features
Clinical manifestations include subacute or chronic dyspnea and cough that last an average of 6 months, most of which occur in non-smoking, middle-aged women. Bilateral end-inspiratory crackles at the base of chest can be heard but most of physical findings are nonspecific. Pulmonary function tests show a restrictive ventilatory defect2. Because similar clinical manifestations can occur in the patients with HP, drug toxicities and occupational lung diseases, detailed history taking is necessary to find exposure to specific antigens such as birds, drugs, or occupational exposures. In addition, the NSIP pattern is known to be the most common histologic pattern of pulmonary involvement in various CTDs. Therefore, in order to exclude specific CTDs, it is important to check for symptoms such as Raynaud's phenomenon, arthralgia or arthritis, skin rash, dry mouth, and dry eye. Autoantibodies, such as anti-nuclear antibody, rheumatoid factor, and anti-cyclic citrullinated peptide, are also useful markers and should be considered for exclude CTDs8. However, in many patients, even if autoantibody is positive, clinical manifestations are not sufficient for diagnosis of specific CTD2,9,10,11,12. If the diagnostic criteria of a specific CTD are not met despite autoantibodies or some clinical features are similar to those of CTD, diagnosis as an idiopathic NSIP can be made. Recently, there has been a proposal to use the term ŌĆ£interstitial pneumonia with autoimmune features (IPAF).ŌĆØ IPAF is a term to characterize the heterogenous group of patients with IIPs who have a clinical flavor of underlying CTDs but do not meet the current American College of Rheumatology criteria for CTDs13. But, the concepts of IPAF are proposed to provide a platform for the prospective study of these heterogenous group and are not intended as guidelines for clinical care. At present, IPAF will need to be validated via prospective research studies and we should be careful to apply it to real clinical field.
2. Diagnosis
The diagnosis of NSIP is similar to that of other interstitial pneumonias including IPF, and it is important to diagnose it through multidisciplinary diagnosis (MDD) with respiratory physicians, radiologists, and pathologists. MDD is particularly important to diagnose idiopathic NSIP because NSIP can occur in various clinical diseases including HP, CTD, and drug reactions1. The differential diagnosis is summarized in Table 1.
1) Radiological examination
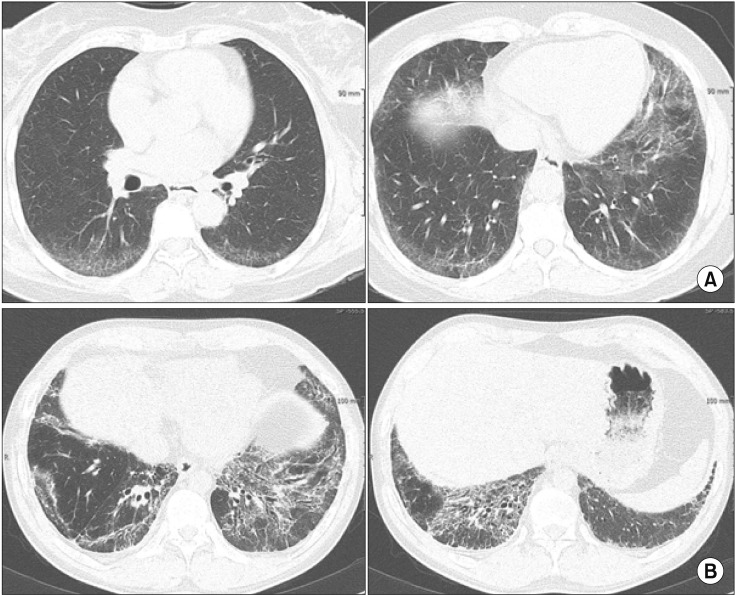
The common findings in thoracic high-resolution computed tomography (HRCT) in NSIP are bilateral reticular opacities, traction bronchiectasis, reduced volume of the lobes, and ground-glass opacity in the lower lungs2,14,15,16. These lesions can involve diffuse bilateral lungs or subpleural area. Although it is not common, 20% of patients showed subpleural sparing which is helpful in distinguishing NSIP from IPF. Unlike UIP, honeycombing is sparse or absent (Figure 1). Consolidation, if present, indicates organizing pneumonia component and may suggest the presence of CTD2. Unlike UIP, it is not possible to diagnose NSIP with HRCT findings alone.
2) Bronchoscopy
Although T lymphocyte fractions increase (>20%) in the bronchoalveolar lavage (BAL) fluid of most patients, it is nonspecific finding and only plays an auxiliary role for differential diagnosis. For example, increase of T lymphocyte fractions in BAL fluid could also be present in HP. If there is no lymphocytosis in the BAL fluid and the fraction of the neutrophil is high, the possibility of IPF rather than NSIP should be considered17,18. Transbronchial lung biopsy is not recommended for definite diagnosis of NSIP because the size of specimen is insufficient.
3) Lung biopsy
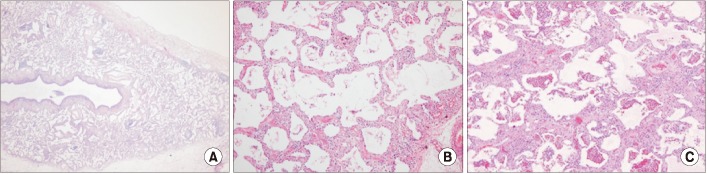
Lung biopsy is essential for definite diagnosis of NSIP. The histologic features of NSIP pattern include diffuse interstitial inflammation and fibrosis which are temporally homogeneous. And, usually the basic structure of the alveoli is preserved (Figure 2)2,3. NSIP pattern can be classified into cellular and fibrotic NSIP. In cellular NSIP, chronic inflammatory cells infiltrate the alveolar wall and fibrosis of the alveolar wall is hardly observed. Fibrotic NSIP is associated with alveolar wall thickening and fibrosis with or without infiltration of inflammatory cells in the alveolar wall5,19,20. Fibrotic NSIP is more common than cellular NSIP and accounts for 80%-90% of the total NSIP2,21. The organizing pneumonia or honeycombing should be sparse or absent. The extent of organizing pneumonia is less than 10% to 20%, even if observed (Table 2).
The diagnosis of idiopathic NSIP can be summarized as the following.
Treatment
1. PICO for treatment of patients with idiopathic NSIP
PICO1. Is steroid treatment effective in patients with idiopathic NSIP?
PICO2. Is combination therapy with steroid and immunosuppressive agent more effective than steroid monotherapy in patients with idiopathic NSIP?
1) Recommendations
- We suggest that clinicians use steroids for treatment of idiopathic NSIP (very low certainty of evidence, strong recommendation).
- We suggest that clinicians use combination with steroids and immunosuppressive agents if steroid monotherapy is ineffective (very low certainty of evidence, weak recommendation).
Idiopathic NSIP is usually treated with steroids and immunosuppressive agents. However, considering the side effects of the mediations, careful observation without treatment can be possible if the disease is mild and meticulous follow up of disease progression with symptoms, pulmonary function and chest HRCT are guaranteed22. Because idiopathic NSIP has a low prevalence and has relatively recently been categorized as an independent disease entity, there are no randomized controlled trials that have demonstrated the natural remission rate without treatment or the therapeutic effect of medications. However, previous retrospective studies have reported that steroids and other immunosuppressive agents induce remission or stabilization of symptoms and pulmonary function23,24. Kim et al.24 reported that among 35 patients with histologically confirmed NSIP who were treated with prednisolone (0.5 mg/kg), 32 patients survived and pulmonary function improved in 24 patients and maintained stable conditions in 6 patients. In study of Watanabe et al.25, after 1 year of steroid treatment, all of 10 patients showed improvement in lung function and oxygenation and only one patient died after 4.3 years of follow-up.
However, this treatment strategy with steroids and immunosuppressive agents has its limitation. It is effective when the inflammatory mechanism is predominant, such as cellular NSIP or organizing pneumonia, but ineffective in patients with fibrotic NSIP26. Recently, opinions, that anti-fibrotic drugs used in IPF may be helpful in these fibrotic subtypes, has been raised, but further studies are required22.
There is no clear guideline of dose and duration of steroids, but it is recommended that 0.5 to 1.0 mg/kg or 40 to 60 mg of prednisone can be used as the initial dose. The dose should be maintained for 1 month, and then slowly be reduced23,26. If the disease is severe, high-dose methylprednisolone therapy (1 g/day for 3 days, followed by 1 mg/kg orally, then gradually reduced) may be performed27,28.
To make this guideline, the authors attempted a systematic review and meta-analysis of the steroid effect in idiopathic NSIP, but all studies were retrospective and comparative analysis was not possible due to the lack of control group. However, according to Xu et al.29, 17 patients (22.9%) died during the 54┬▒34-month follow-up period and 34 patients (45.9%) died during steroid withdrawal in 74 patients with idiopathic NSIP, which proved the effect of steroids. Park et al.23 reported that the mean duration of steroid treatment in NSIP was 17.4┬▒12.1 months and 36% of the patients experienced recurrence. Kim et al.24 reported recurrence in six out of 30 steroid-treated patients (20%), which was associated with an initial low dose (0.5 mg/kg) of steroid and short treatment duration.
Immunosuppressive agents include azathioprine, cyclophosphamide, cyclosporine, and mycophenolate mofetil, which are used for steroid sparing or enhancing the steroid effect. There is no consensus on whether these agents should be initiated at the time of diagnosis or the time of disease progression or the occurrence of steroid dependence. In general, these medications are used when there is a dependence on steroids or the need of steroid maintenance for preventing disease recurrence26,30.
Among these immunosuppressive agents, cyclophosphamide is the most frequently used. Kondoh et al.31 conducted a study in which they treated 12 histologically confirmed fibrotic NSIP patients with combination of low-dose steroids (20 mg prednisone, every other day) and cyclophosphamide (1 to 2 mg/kg/day). As a results, 33% showed improvement, 67% (8/12) showed stable response, and 21% had side effects related to cyclophosphamide (hemorrhagic cystitis, leukopenia, myelodysplastic syndrome, and infection)31. Corte et al.32 reviewed 54 patients with rapid progression of NSIP who were treated with IV cyclophosphamide (600 mg/m2) and observed good response in most of the patients after 6 months of treatment. Effects of other drugs such as azathioprine, cyclosporine, and mycophenolate mofetil have been reported in a small number of cases4.
On the other hand, literature review was conducted to confirm whether the combination of steroids and immunosuppressive agents is more effective than steroid monotherapy. In a study published by Fujita et al.10, 22 patients with idiopathic NSIP were treated (19 with steroids and 3 with steroids and cyclophosphamide), and the mortality rate of patients with combination therapy was 1.5 times (95% confidence interval, 0.26-9.79) that of the patients with steroid monotherapy. However, the study was a small, retrospective study and the patients who were more severe or unresponsive to steroids may have treated with combination therapy. Therefore, the results of this study were needed to be interpreted with caution. Recently, Keir et al.33 reported the hopeful results with rituximab in 50 patients with severe interstitial lung disease who did not respond to conventional immunosuppressive agents except IPF. In the future, studies on the efficacy of these drugs in idiopathic NSIP need to be conducted.
There is no standardized method for assessing therapeutic response in idiopathic NSIP. However, like IPF, treatment response can be measured by combining the changes of HRCT, symptoms such as dyspnea or cough and pulmonary function test (forced vital capacity [FVC] and carbon monoxide diffusion capacity [DLco]), usually between 3 and 6 months after the start of treatment34. Changes in pulmonary function over time are known to be useful surrogate marker of survival. Reduction of DLco over 15% after 12 months or decrease of FVC over 10% between 6 and 12 months are independently associated with mortality23,35,36. Therefore, when DLco is reduced over 15% or FVC is reduced over 10% despite treatment without any specific reason such as infection, other treatments such as changing medications or lung transplantation should be considered37. In addition to pharmacologic treatment, oxygen therapy for hypoxemia at night or during exercise, treatment for concomitant diseases such as reflux esophagitis or pulmonary hypertension, and symptomatic therapy for dyspnea or cough can be performed38.
Natural Course and Prognosis
Survival of NSIP is better than IPF39. Many studies have reported a 5-year survival rate of more than 70%, and particularly in the case of cellular NSIP, there are few disease-related deaths2,7,23. Treatment of idiopathic NSIP, although not well proven, is generally begin with steroid only or combined with immunosuppressants such as azathioprine, cyclophosphamide, cyclosporine, and mycophenolate mofetil. The clinical response after the first treatment was generally favorable. About two thirds of patients have improved after initial treatment.
However, frequent recurrences were reported after discontinuation of treatment. Kim et al.24 reported recurrence in 20% and Park et al.23 reported recurrence in 36%, and the prognosis of patients with recurrence was worse. In addition, as in the IPF, there may be acute exacerbation during the course of NSIP, and Park et al.40 reported acute exacerbation of 4.2% for one year in patients with idiopathic NSIP.
NSIP pattern is the most common interstitial pneumonia that occurs in CTDs. Therefore, it has always been suggested that NSIP may be the first or the only manifestation of CTD. The possibility that NSIP could be the first manifestation of CTD was firstly raised by Sato et al.41. Kinder et al.11 reported that 80% of 28 idiopathic NSIPs met the criteria for undifferentiated connective tissue disease (UCTD), and Park et al.23 reported that eight of 83 patients (10%) initially thought to be idiopathic NSIP were diagnosed with CTD in the process of their illness. In fact, a large number of patients with NSIP currently do not meet the criteria for specific CTD have some features of CTD. The researchers described these patients with other names, such as UCTD-interstitial lung disease, lung-dominant CTD, or autoimmune featured ILD. Recently, ATS/ERS proposed ŌĆ£interstitial pneumonia with autoimmune features (IPAF)ŌĆØ for them42. Therefore, patients with histologically NSIP pattern should go through sufficient consideration of the presence of CTD, and the occurrence of CTD should be carefully observed in the following process.
 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Print
Print Download Citation
Download Citation