Introduction
Autophagy is a defense mechanism to protect cells from multiple stresses such as nutrient starvation, hypoxia, pathogen infection, and radiation1. During the initial stages of the autophagic process, long-lived cellular proteins and organelles are sequestered and engulfed by intracellular double-membrane-bound structures called autophagosomes (early autophagic vesicles)2. Autophagosomes mature by fusing with lysosomes to form autolysosomes (late autophagic vesicles), in which the sequestered proteins and organelles are digested by lysosomal hydrolases and recycled to sustain cellular metabolism3. In addition, autophagy is observed in cancer cells upon treatment with a wide spectrum of cytotoxic and targeted chemotherapeutic agents3-5. Thus, concurrent autophagy inhibition can be expected to have a synergistic effect with chemotherapy on cancer cell death4.
Monensin, a polyether antibiotic, is known as an autophagy inhibitor, which interferes with the fusion of autophagosome and lysosome and thus blocks the autophagic process at its final step6. Used alone, its cytotoxic effect is negligible and there have been few reports of its effect in combination with standard anticancer drugs5,6.
The epidermal growth factor receptor (EGFR) is a member of the receptor tyrosine kinase family, which is important in cancer cell growth, proliferation, invasion, and metastasis7. It is frequently deregulated in non-small cell lung cancer (NSCLC)8. Tyrosine kinase inhibitors such as gefitinib and erlotinib have been developed for the treatment of NSCLC, through inhibition of the tyrosine kinase activity of EGFR9. Gefitinib and erlotinib are effective in NSCLC cells harboring activating EGFR mutations, but not in cells with wild type EGFR10,11.
The phosphoinositide 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway is activated in many cancers, including NSCLC12,13. This signaling pathway plays an important role in cell growth, cell proliferation, angiogenesis, and protein synthesis14. Inhibitors of mTOR, including rapamycin, have demonstrated efficacy in vitro and are now being tested in early-phase clinical trials in NSCLC13.
In this study, we investigated whether erlotinib, an EGFR inhibitor, or rapamycin, an mTOR inhibitor, is effective in combination therapy with monensin in NSCLC cells. We also explored the mechanisms of the combination effect, and demonstrated that the inhibition of autophagy with monensin enhances rapamycin- and erlotinib-induced apoptotic cell death and cell cycle arrest.
Materials and Methods
1. Cell cultures
NCI-H1299 cells, which are derived from NSCLC and lack EGFR mutations, were purchased from the American Type Culture Collection (Rockville, MD, USA). NCI-H1299 cells were maintained as monolayer cultures in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, and 1% sodium pyruvate, at 37℃ in a humidified incubator under 5% CO2 gas. All cell culture materials were obtained from Welgen (Daegu, Korea).
2. Reagents
Acridine orange (AO), dimethyl sulfoxide (DMSO), propidium iodide (PI), 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) solution, and anti-β-actin antibody were purchased from Sigma (St. Louis, MO, USA). Monensin, rapamycin, and erlotinib were purchased from Selleck (Houston, TX, USA). The Annexin V-FITC kit and anti-p21 antibody were obtained from BD Biosciences (San Jose, CA, USA). Antibodies against phospho-p70S6K, LC3, caspase-3, cleaved-caspase 3, poly(ADP-ribose) polymerase (PARP), bcl-2, and notch3 were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies against bax, bcl-xl, skp2, and p27 and horseradish peroxidase (HRP)-conjugated secondary antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The enhanced chemiluminescence (ECL) Western blotting detection system was supplied by Amersham Biosciences (Piscataway, NJ, USA).
3. Treatments
NCI-H1299 cells were seeded in a 96-well plate (1.2×103 cells/50 µL) and incubated at 37℃. The next day, cells were pre-treated with 50 nM monensin for 4 hours, followed by treatment with varying concentrations of rapamycin or erlotinib for 48 hours, after which further analysis was performed. Rapamycin (20 mM), erlotinib (40 mM), and monensin (1 mM) were dissolved in DMSO. As a control, equal volumes of DMSO (0.05%) were added to untreated cells.
4. Analysis of cell viability
Inhibition of cell proliferation was determined by a MTT assay. MTT solution was added to cells in 96-well plates to a final concentration of 0.5 mg/mL, and cells were incubated at 37℃ for 4 hours. After removing the culture media, 50 µL of DMSO was added, and the optical density of each well was read at 590 nm.
5. Quantification of acidic vesicular organelles (AVOs) with AO staining
Cells were seeded in 60 mm dishes and treated with rapamycin or erlotinib with or without pretreatment with 50 nM monensin for 4 hours. 48 hours after incubation, AO staining was performed; briefly, the treated cells were stained with AO (1 µg/mL in serum-free RPMI 1640 media) in the dark at 37℃ for 15 minutes, and then washed with serum-free RPMI 1640 media. Images of AO staining were visualized immediately using a Leica confocal laser scanning microscope (Wetzlar, Germany).
6. Annexin V-FITC assay by flow cytometry
After incubation with each agent, cells were washed with phosphate buffered saline (PBS), trypsinized, collected in a 15 mL conical tube, and pelleted by centrifugation (1,200 rpm) for 10 minutes at room temperature. The pellets were washed twice with PBS, and then resuspended in annexin V binding buffer (150 mM NaCl, 18 mM CaCl2, 10 nM HEPES, 5 mM KCl, 1 mM MgCl2). Fifteen minutes after incubation with annexin V (1 g/mL) in the dark, the cells were analyzed using a FACS-Canto II flow cytometer (BD, San Jose, CA, USA).
7. DNA content analysis by flow cytometry
After incubation, cells were washed with ice-cold PBS, tryp-sinized, collected in a 15 mL conical tube, and pelleted by centrifugation (1,200 rpm) for 10 minutes at 4℃. The pellets were washed twice with ice-cold PBS, fixed in 70% ethanol, washed in PBS, resuspended in 300 µL of PBS containing 50 µg/mL PI and 50 µg/mL RNase A, and incubated in the dark for 15 minutes at room temperature. The DNA content of each cell nucleus was determined by flow cytometry.
8. Western blot analysis
Thirty micrograms of protein were resolved by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membranes were blocked with 5% skim milk-PBS-0.1% Tween 20 for an hour at room temperature before being incubated overnight with primary antibodies diluted 1:1,000 in 5% skim milk-PBS-0.1% Tween 20. The membranes were then washed three times in 1× PBS-0.1% Tween 20 and incubated with HRP-conjugated secondary antibodies diluted 1:2,000 in 5% skim milk-PBS-0.1% Tween 20 for an hour. After successive washes, the membranes were developed using an ECL kit.
9. Analysis of combination effects
For the statistical analysis of the synergistic effect of drug co-administration on MTT analysis, combination indexes (CI) were calculated using CalcuSyn software version 2.1 (Biosoft, Cambridge, UK). CI<0.7 indicates synergistic effects (the smaller the value, the stronger the synergism); CI=1 indicates additive effects; and CI values>1 suggest antagonistic effects.
Results
1. Growth inhibition induced by rapamycin or erlotinib is enhanced by combination treatment with monensin in NCI-H1299 cells
We examined the cell growth inhibition of rapamycin or erlotinib when each drug was combined with monensin, an autophagy inhibitor. NCI-H1299 cells were pretreated with monensin (50 nM) for 4 hours and then treated with various concentrations (0.20 µM) of rapamycin or erlotinib for 48 hours. Figure 1A shows that both rapamycin- and erlotinib-induced morphological change and cell growth inhibition, observed by light microscopy, are increased by adding treatment with monensin. Cell viability was also measured using the MTT assay (Figure 1B). Treatment with each drug alone causes a slight decrease in cell proliferation; by contrast, this decrease is significantly enhanced when cells are treated in combination with monensin. To assess whether this combination effect is synergistic or additive, we calculated the CI at each concentration (2.5-20 µM). As shown in Figure 1C, the CI values of all combinations tested are below 0.7, indicating that the agents act synergistically when used as combination treatments in NCI-H1299 cells.
2. Autophagy inhibition by monensin enhances apoptosis induced by rapamycin or erlotinib in NCI-H1299 cells
We investigated whether the observed changes in growth inhibition induced by monensin were due to enhanced apoptosis. First, we performed AO staining to detect the AVOs formed during the process of autophagy (Figure 2A). Both rapamycin- and erlotinib-treated cells show plentiful orange staining, but cotreatment with monensin (50 nM), which inhibits fusion of autophagosomes with lysosomes, effectively suppresses the development of AVOs in NCI-H1299 cells. Then we performed Annexin V-FITC assays by flow cytometry. As shown in Figure 2B, monensin cotreatment increases annexin V-positive apoptotic cells to about 20% compared with treatment using rapamycin or erlotinib alone. We also analyzed the sub-G1 cell population, characteristic of apoptosis, by flow cytometry with PI staining (Figure 2C). The sub-G1 cell population increases significantly when monensin is used in combination treatment with rapamycin (3.62-20.60%) or erlotinib (4.25-22.66%), compared to treatment with either agent alone. These results indicate that the observed combination effect is caused by an increase in apoptosis.
3. The combination effect of monensin with rapamycin or erlotinib is mediated by apoptotic signaling
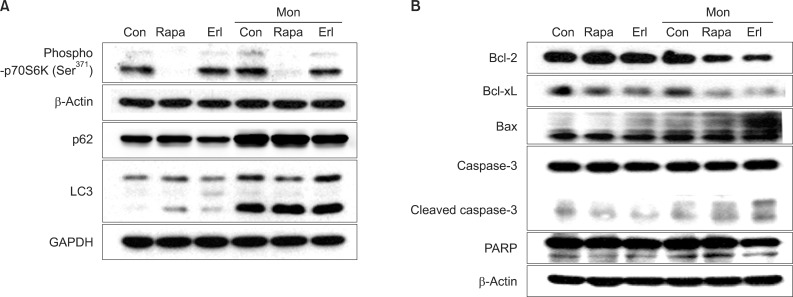
We further examined the expression of proteins related to apoptosis during this process in NCI-H1299 cells. First, we confirmed that phosphorylation of p70S6K (a downstream target of mTOR) at Ser371 is substantially decreased by rapamycin treatment and that type II LC3 (14 kDa), a hallmark of the autophagosome, is remarkably increased and degradation of p62 (an adaptor protein that binds to LC3 and is removed by autophagy) is blocked by monensin treatment, indicating the accumulation of autophagosomes (Figure 3A). When compared to treatment with rapamycin or erlotinib alone, cotreatment with monensin induces increased levels of pro-apoptotic proteins, including Bax, cleaved caspase 3 and cleaved PARP, and decreases the levels of anti-apoptotic proteins, including Bcl-2 and Bcl-xL (Figure 3B).
4. Combination treatment with monensin increases cells arrested in G1-phase
We also analyzed the effect of monensin treatment on cell cycle progression by flow cytometry with PI staining (Figure 4A). DNA content analysis shows that cotreatment with monensin increases the numbers of cells in the G1 cycle phase, as compared to treatment with rapamycin or erlotinib alone. Next, we investigated the effect of monensin on cell cycle regulators, including notch3, skp2, p21, and p27 by western blot analysis (Figure 4B). Notch signaling is known to induce skp2 expression and promote reduction of p21 and p27 cyclin-dependent kinase inhibitors. Expression of notch3 and skp2 is decreased after treatment with monensin. In contrast, p21 expression is noticeably increased by monensin treatment. This result is in agreement with the increased G1 cycle arrest caused by monensin treatment. The levels of p27 are unchanged after each treatment.
Discussion
It is known that the predominant role of autophagy in cancer cells is to confer stress tolerance, which serves to maintain tumor cell survival1. Cytotoxic and metabolic stresses, including hypoxia and nutrient deprivation, can activate autophagy for recycling of ATP to maintain cellular biosynthesis and survival15. In cancer cells that survive chemotherapy or radiation, activation of autophagy may enable a state of dormancy in residual cancer cells that may contribute to tumor recurrence and progression4. Inhibition of autophagy in tumor cells has been shown to enhance the efficacy of anticancer drugs, supporting its role in cytoprotection16.
Multiple studies have shown that genetic knockdown of autophagy-related genes (Atgs) or pharmacological inhibition of autophagy can effectively enhance tumor cell death induced by diverse anticancer drugs in preclinical models15,17. For example, inhibition of autophagy improved responses to alkylating agents in tumor cells18,19. In apoptosis-defective leukemic and colon cancer cell lines, inhibition of autophagy was shown to sensitize resistant cells to tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis20. Furthermore, inhibition of autophagy enhanced apoptosis induction by cetuximab, an antibody against EGFR21. However, the effect of autophagy inhibitors on lung cancer chemotherapy is not well documented, and therefore, it was addressed in this study.
Pharmacological inhibitors of autophagy can be broadly classified as early- or late-stage inhibitors of the pathway. Early-stage inhibitors include 3-methyladenine22, wortmannin23, and LY29400224, which target class III PI3K and interfere with its recruitment to membranes. Late-stage inhibitors include chloroquine25, hydroxychloroquine26, bafilomycin A127, and monensin6. Bafilomycin A1 is a specific inhibitor of vacuolar-ATPase, and monensin and chloroquine/hydroxychloroquine are lysosomotropic drugs that prevent the acidification of lysosomes, whose digestive hydrolases depend on low pH. Multiple clinical trials are currently assessing the effects of combined treatments with various anti-cancer drugs plus hydroxychloroquine for patients with various refractory malignancies28. Monensin is a monocarboxylic acid ionophore drug isolated from Streptomyces cinnamonensis; it can form complexes with metal cations and is able to transport them through cellular and subcellular membranes6. Monensin also blocks intracellular protein transport and exhibits antibiotic, antimalarial, and other biological activities29. It has an excellent safety record as an additive to beef cattle feed; however, its anticancer effect as an autophagy inhibitor has not been established30.
In this study, we showed that a nanomolar concentration (50 nM) of monensin can enhance apoptosis induced by rapamycin or erlotinib in NCI-H1299 cells. To the best of our knowledge, this is the first report showing that autophagy inhibition with monensin can enhance the effect of anticancer agents in lung cancer cells. Increased levels of bax, cleaved caspase 3, and cleaved PARP accompany this process. Monensin can also induce accumulation of cells in the G1 phase by modulating cell cycle regulators. Expression of notch3 and skp2 is decreased but p21 expression increases by monensin treatment. Notch3 and skp2 are positive regulators of cell cycle progression, whereas p21, a cyclin dependent kinase inhibitor, is a negative regulator that causes cell cycle arrest by preventing interaction between cyclins and cyclin-dependent kinases.
Taken together, our data suggest a strategy to enhance the effects of EGFR tyrosine kinase inhibitors and mTOR inhibitors by blocking autophagy, resulting in increased growth inhibition and apoptotic cell death of NSCLC cells.



 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Print
Print Download Citation
Download Citation