Introduction
Chronic obstructive pulmonary disease (COPD) is a heterogeneous disease characterized by chronic respiratory symptoms accompanied by persistent airflow limitation [1]. Emphysematous changes in the lung are frequently observed in patients with COPD because of alveolar wall destruction due to toxic inhalation (e.g., cigarette smoke), causing distal airspace enlargement [2,3]. Consequently, maximal expiratory airflow is decreased by loss of elastic recoil of emphysematous lung [3]. Inflammation and peribronchial fibrosis in small conducting airways (<2 mm in diameter), referred to as small airway disease (SAD), are also major pathologic changes in COPD [3]. Additionally, interstitial lung disease (ILD) is a cluster of diseases characterized by inflammatory and fibrotic infiltration of alveolar septa that cause substantial changes in the alveolar epithelium and capillary endothelium [3]. Both COPD and ILD have distinct features, but may coexist in a single patient because they share some similar risk factors (e.g., old age, male sex, and tobacco exposure) [1,4-8]. In this review, we discuss current issues regarding the clinical view of ILD in patients with COPD with respect to prevalence, prognosis, pathophysiology, management, and pharmacologic treatments.
Prevalence
Cottin et al. [9] first proposed the disease entity “combined pulmonary fibrosis and emphysema” (CPFE), which was defined as diffuse interstitial fibrosis along with emphysematous changes in upper lobes. Interstitial changes vary in patients with CPFE, such that most are unclassifiable, one-third have the usual interstitial pneumonia pattern, and one-fifth constituted classifiable [10]. Among classifiable ILD, connective tissue diseases (CTD)-ILD composites nearly half of the patients and other classifiable ILD including idiopathic non-specific interstitial pneumonia, eosinophilic pneumonia, chronic hypersensitivity pneumonia, desquamative interstitial pneumonia/respiratory bronchiolitis-ILD composite the other half. In a multicenter retrospective study analyzing 34 CPFE patients with CTD-ILD (CTD-CPFE), 53% of patients had rheumatoid arthritis (RA), 29% had systemic sclerosis, 23% had mixed or overlap CTD and 12% had other CTDs [11]. A pooled prevalence of CTD-ILD associated with systemic sclerosis in three previous studies was reported to be 13.4% [12-15]. The prevalence of emphysema (>5%) in high-resolution computed tomography (HRCT) was reportedly 13.4%-25.4% and COPD was evident in 9.2% of patients with idiopathic pulmonary fibrosis (IPF) [16-18]. Furthermore, among patients with RA-related ILD, emphysema was found in 27% of non-smokers and 76% of smokers [19].
Interstitial lung abnormalities (ILA) are defined as increased lung densities on chest computed tomography (CT) images of patients without previous history of ILD [20-22]. Because these changes in the lungs have similar but milder clinical characteristics than ILD, they are presumed to be early or mild manifestations of ILD [20]. These changes reportedly coexist in 13.5% of patients with COPD [23].
Pathophysiology and Biomarkers
The pathogenesis of CPFE has been still largely unknown, and previous studies have suggested possible pathophysiology of the disease (Figure 1). Because IPF and emphysema share similar risk factors (e.g., cigarette smoking), simultaneous or successive emphysema and fibrosis may develop. In patients with simultaneous development, the locations of emphysema and fibrosis may be distributed in similar regions of the lung [24]. Basal lung fibrosis has also been suggested to proceed initially along upper lung field tracts, thereby promoting the progression of emphysema [9]. Some studies have speculated that autoimmune features or gastroesophageal reflux disease caused by smoking habits may affect CPFE progression [5,25,26].
Some genetic backgrounds have been studied as potential predisposing factors for CPFE progression [27,28]. There have been reported that some genes, such as FAM13A and TERT gene, are associated with both COPD and IPF progression [29-32]. A recent study conducted by Guzman-Vargas et al. [33] investigated the association of several genes, including FAM13A, TERT, DSP, and TOLLIP, which are reported to be related to IPF progression [33]. The study has shown that rs2736100C allele in TERT gene and specific genotype of rs2076295 (DSP) are associated with increased risk of CPFE. The ATP-binding cassette subfamily A member 3 (ABCA3) and surfactant protein-C (SFTPC) genes have important roles in the maintenance of lung surfactant homeostasis, and a few case reports have implied associations between CPFE and mutations in those genes [34,35]. A recent study by Kinjo et al. [36] investigated the Gly82Ser mutation in the advanced glycosylation end-product specific receptor (AGER) gene in 111 patients with CPFE and 337 patients with COPD in Japanese patients. It revealed that the AGER gene mutation leads to receptor for advanced glycation end products pathway inhibition, which may be associated with lung fibrosis in patients with CPFE.
Efforts to reveal immunological and inflammatory mechanisms of CPFE progression have included several studies of biomarkers associated with the disease. Cornwell et al. [37] studied explanted lung tissue from patients who died of lethal trauma. They categorized lung tissue into emphysema only, IPF, CPFE, and healthy control groups, then compared levels of 34 inflammatory biomarkers [37]. Regional comparisons of biomarkers showed no significant differences among groups (i.e., emphysema, IPF, or CPFE), except in the level of CCL22. Additional comparisons of biomarkers between groups revealed four clear categories: biomarkers elevated in emphysema alone (interleukin [IL]-6, CXCL10, CCL2, IL-10, and interferon-γ); biomarkers reduced in emphysema alone (IL-17A, IL-13, matrix metalloproteinase [MMP] 1, MMP13, MMP2, CCL24, and lysyl oxidase); biomarkers that were similar among emphysema, IPF, and CPFE, but different from the healthy control group (sCD14, CCL5, tumor necrosis factor [TNF] RII, SP-D, ST2, IL-1β, IL-4, IL-5, CXCL8, and TNFα); and biomarkers with a mixed pattern that did not fit any other categories. These results suggest that the levels of inflammatory proteins are generally comparable between IPF and CPFE, but distinct features are present in emphysema. These results were also consistent with the findings of previous studies [38-41]. Therefore, the immunological pathogenesis of CPFE may be more closely associated with IPF development.
Klebs von den Lungen-6 (KL-6) is a high-molecular-weight glycoprotein that is strongly expressed on type II alveolar pneumocytes and bronchiolar epithelial cells. It is an important marker of ILD and is associated with disease severity, progression, and survival; notably, it promotes pulmonary fibroblast migration and proliferation [42-45]. Because of this association with ILD, some studies have investigated relationships with fibrotic changes in patients with COPD. In asthma-COPD overlap, patients with a history of smoking and older age have more frequent ILA [46]. These patients with asthma-COPD overlap and ILA showed higher levels of KL-6, compared with patients with asthma-COPD overlap who lacked ILA. The clinical significance of KL-6 has also been investigated in patients with CPFE. Chiba et al. [41] revealed that KL-6 and SP-D were both correlated with lung volume and composite physiologic index among patients with CPFE. Furthermore, a high level of KL-6 in patients with CPFE is reportedly a strong predictor of frequent acute exacerbation of IPF (AE-IPF), similar to its role in patients with IPF [47,48]. These results indicate that KL-6 is an important biomarker related to fibrotic changes and disease prognosis in patients with CPFE.
Radiologic Features
HRCT is considered to be the most important diagnostic tool of CPFE, as its definition is based on radiologic features. Although it was initially suggested as coincidence of upper lung emphysema with features of diffuse interstitial lung fibrosis, consecutive studies have used this term heterogeneously (Figure 2) [9,49]. In total of 72 studies with CPFE patients, 60% of studies have defined CPFE with emphysema located in the upper zone, and 40% with emphysema in all location or did not specify its location [49]. Also, 72% of studies have enrolled patients with emphysema by any extent, but 28% of studies have limited by specific threshold, including emphysema presented ≥10% of lung volume (9 studies), ≥5% (4 studies), and ≥25% (4 studies). Majority of the studies (97%) have evaluated the extent of lung fibrosis visually.
Emphysematous lesions of CPFE can be presented as centrilobular, paraseptal or mixed lesion of both type. Among these manifestations, there were discrepancies between studies on majority type of emphysema; centrilobular [50-52], paraseptal [9,53-55] or mixed type [10,56] were reported to be most common type of emphysema in different studies. Regarding fibrotic area of CPFE, usual interstitial pneumonia has been shown to be the most common histological morphology in previous studies [9,57]. Honeycombing, reticular opacities, traction bronchiectasis and ground-glass opacities were most frequently seen in HRCT findings [9]. Distribution pattern of CPFE were described as three patterns: (1) area of emphysema and fibrosis distributed in completely separated pattern (emphysema positioned in the upper zones and fibrotic area in the basal zones), (2) progressive transition of diffuse upper zone emphysema to a zone of transition between bullae and honeycombing, (3) paraseptal emphysema positioned within fibrotic areas [58].
Thick-walled large cysts (TWLCs), which were defined as cysts with diameter ≥2 cm surrounded by a wall ≥1 mm thick, were proposed as a unique feature of CPFE [11,55]. Inomata et al. [55] performed an autopsy study of 22 CPFE patients in comparison with 8 IPF and 17 emphysema-alone patients. TWLCs were only presented in CPFE patients, and neither IPF nor emphysema-alone patients showed this lesion [55]. In this study, total of 72.7% patients were shown to have TWLCs in CPFE patients, radiologically and pathologically (68.8% in upper lobes and 62.5% in lower lobes, radiologically). TWLCs were found in the centriacinar/centrilobular lesions, involving one or more acini, membranous and respiratory bronchioles, with alveolar destruction, dense wall fibrosis and occasional fibroblastic foci. These lesions were shown to be presented belong upper paraseptal emphysema near chest wall or shown within zone of fibrosis [59]. It may be caused by development of lung fibrosis within emphysematous lesions, and expanded by retraction induced by fibrotic process [28,58]. Although TWLCs are exclusively presented rather than IPF or emphysema-alone patients, differential diagnosis including lung abscess, cancer, mycobacterial/fungal infections should be considered [60].
Prognosis
1. Prognosis of CPFE
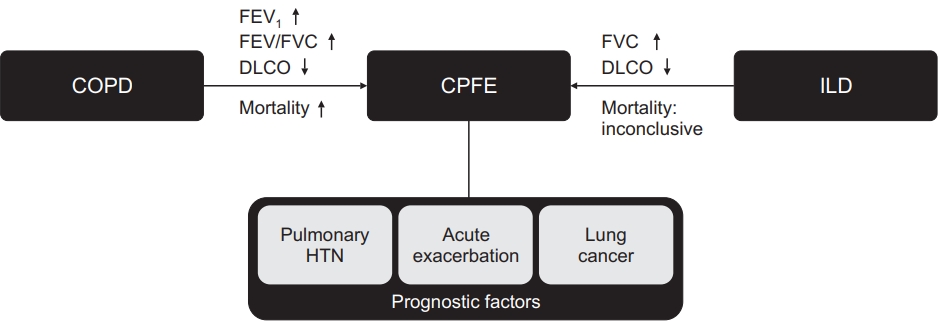
Although there are no exact diagnostic criteria including the extent or localization of emphysema, the clinical implications of CPFE (determined using various definitions) are reportedly substantial [49,61]. A previous study reported that 5-year survival was 54.6% and median survival was 6.1 years in patients with CPFE [9]. Notably, accompanying pulmonary fibrosis in patients with emphysema resulted in five-fold greater mortality risk, compared with nonmalignant patients who did not have pulmonary fibrosis (Figure 3) [50]. However, there are conflicting results regarding mortality in patients with CPFE and patients with IPF alone: some studies reported no significant difference in mortality [16,17,62-64], while others reported that mortality was higher in CPFE groups [56,65-68] or lower in CPFE groups [69,70]. In a meta-analysis performed by Koo et al. [12], higher mortality was noted in patients with CTD-CPFE, compared to those without CTD-ILD with emphysema [12]. Various studies that analyzed factors associated with CPFE was shown in Table 1.
The most important factors that influence CPFE prognosis are acute exacerbation, pulmonary hypertension, and the presence of lung cancer [5,66,71,72] (Figure 3). Previous studies have reported that the prognosis of acute exacerbation is favorable, and that acute exacerbation is less frequent in patients with CPFE than in patients with IPF. However, it may have lethal consequences and careful monitoring is needed [73,74]. Zantah et al. [74] stratified exacerbation events in patients with CPFE into acute exacerbation of COPD (AE-COPD) and AE-IPF. AE-COPD comprised clinical symptoms of airway obstruction with radiologic findings such as airway wall thickening, mucus impaction, atelectasis, consolidation, and mediastinal adenopathy. That study only included severe exacerbation, which was defined by events that led to hospitalization, as defined in the Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2018 guideline. By contrast, AE-IPF comprised acute respiratory deterioration that led to hospitalization and the use of high-dose corticosteroids. Furthermore, it involved radiologic findings of new lesions (e.g., ground-glass opacities, interlobular septal thickening, and consolidation). The authors analyzed the prevalence and prognosis of exacerbation events, as well as the applications of mechanical ventilation or extracorporeal membrane oxygenation. Exacerbation was more prevalent as AE-IPF, rather than AE-COPD (14.1% vs. 11.7%, p<0.01), and all mortalities were related to AE-IPF. Applications of mechanical ventilation or extracorporeal membrane oxygenation were more frequently performed in patients with AE-IPF than in patients with AE-COPD. Therefore, in patients with CPFE who exhibit clinical signs of exacerbation, identification of exacerbation type may be important because AE-IPF requires closer observation and earlier intervention.
Previous studies have shown that pulmonary hypertension is an important poor prognostic factor for CPFE. In a pivotal study, Cottin et al. [9] performed survival analysis of 61 patients with CPFE. They showed that the presence of pulmonary hypertension was significantly associated with poor survival (hazard ratio [HR], 4.03; p=0.03). Patients with pulmonary hypertension at diagnosis had a mean survival of 4.8 years and 5-year survival of 25%, whereas patients without pulmonary hypertension had a mean survival of 9.1 years and 5-year survival of 75%. In another study, Mejia et al. [65] reported that low functional vital capacity (FVC <50% predicted; HR, 2.6; p=0.02) and severe pulmonary hypertension (systolic pulmonary artery pressure ≥75 mm Hg; HR, 2.25; p=0.02) were important clinical factors associated with poor survival. Patients with CPFE who were diagnosed with pulmonary hypertension exhibited 1-year survival of 60% [75]. Overall, pulmonary hypertension is reportedly observed in 29%-68% of patients with CPFE [9,71,75,76].
Because both emphysema and IPF have been identified as independent risk factors for lung cancer, CPFE is presumably a major risk factor for lung cancer development. Furthermore, most patients with CPFE have a history of smoking, and therefore an association may exist between CPFE and lung cancer [63,77]. In a retrospective study of 47 patients with CPFE, 22 patients (46.8%) had concomitant lung cancer [54]. Compared with patients who had emphysema alone, patients with CPFE and patients with IPF had greater risks of lung cancer development (adjusted HR, 4.62 [p<0.01], 4.15 [p=0.046], respectively). The difference between CPFE and IPF groups was not statistically significant [78].
Although lung cancer itself may have severe consequences, it also contributes to poor disease outcomes in patients with CPFE. Oh et al. [79] analyzed risk factors for acute exacerbation in 227 patients with CPFE. They identified the presence of lung cancer (HR, 3.274; p<0.01), as well as sex, age, and physiology (GAP) score (HR, 1.434; p=0.02) as significant risk factors for acute exacerbation. Furthermore, the coexistence of lung cancer is associated with increased mortality. Moon et al. [80] compared patients with CPFE and patients with IPF, all of whom had been diagnosed with non-small cell lung cancer, and found that patients with CPFE had a significantly greater rate of acute exacerbation (HR, 2.26, p=0.029). Furthermore, Usui et al. [81] retrospectively reviewed 1,143 patients with lung cancer to compare clinical outcomes between patients with underlying CPFE and patients with underlying emphysema. The median overall survival of patients with CPFE was 10.8 months, which was worse than the survival of patients with emphysema (21.9 months) or the survival of patients without emphysema or fibrosis (54.0 months).
The prognostic impact of the proportion of abnormal lesions has also been studied in patients with CPFE. Suzuki et al. [51] showed that an increased proportion of abnormal area (i.e., sum of low attenuation area [pixels <-950 Hounsfield units, representing emphysema] and high attenuation area [pixels >-700 Hounsfield units, representing interstitial fibrosis]) in chest CT was significantly associated with frequent hospitalization in patients with CPFE. Nemoto et al. [82] analyzed fibrotic area alone in patients with CPFE, which revealed that patients with a higher proportion of fibrosis had higher mortality, more frequent exacerbations, worse lung function trajectories, and more rapid CPFE progression. Malli et al. [76] analyzed CPFE patients of a Greek cohort, and revealed that ILD extent of more than 30% was associated with poor survival. In contrast, Jacob et al. [85] analyzed 272 IPF patients, and reported that presence or extent of emphysematous area in volumetric chest CT image were not associated with patients’ survival [83]. Thus, revelation of abnormal lesion in CPFE patients is important in predicting the disease prognosis, and use of late gadolinium enhanced thoracic magnetic resonance imaging may have a role in differentiation from pure emphysema [86].
2. Prognosis of ILA in COPD patients
Previous studies have reported that ILA is presented in 8% of smokers and 13.5% of patients with COPD [21,23]. Because it is associated with decreased lung volume [21,87], ILD progression [88], and increased mortality [20,87,88], in combination with a genetic background similar to the background in patients with IPF [89], ILA is presumed to be an earlier or milder form of ILD [49]. Furthermore, ILA patterns are found in considerable numbers of patients with COPD, and thus the clinical significance of ILA has been studied in patients with COPD (Table 1).
Putman et al. [20] analyzed the data of four major cohort studies including the Framingham Heart Study, AGES-Reykjavik study, COPDGene study, and ECLIPSE study. Among 11,691 patients enrolled in these studies, 868 patients (7.4%) had ILA in their CT images. Analyses of all-cause mortality in each cohort showed that ILA was associated with higher mortality (HR, 2.7 [95% confidence interval (CI), 1.1-6.5], 1.3 [95% CI, 1.2-1.4], 1.8 [95% CI, 1.1-2.8], 1.4 [95% CI, 1.1-2.8], respectively). Considering that many patients in the COPDGene study and all patients in the ECLIPSE study had COPD, the presence of ILA is presumably an important risk factor for death in patients with COPD. Additionally, the presence of ILA had a significant effect on mortality, regardless of correction for the volumetric proportion of emphysema.
Ash et al. [22] performed a follow-up study with COPDGene cohort data, which investigated the clinical effects of ILA in patients with COPD. They identified emphysema and ILA features in patients’ chest CT images, using an objective CT analysis tool, then comprehensively analyzed their associations with lung function, health-related quality of life, exercise capacity, and mortality. Compared with patients who had emphysema alone, patients who had both emphysema and ILA demonstrated a higher percent of predicted forced expiratory volume in 1 second (FEV1) and a lower percent predicted diffusing capacity of lung for carbon monoxide (DLCO). However, there were no significant differences in terms of FVC between the two groups. Feldhaus et al. [9]0 also showed that high attenuated volume in quantitative CT was associated with a profound reduction of DLCO. Moreover, Ash et al. [22] showed that exercise capacity (measured with the 6-minute walking test), health-related quality of life (evaluated with St. George’s Respiratory Questionnaire score), and mortality were all unfavorable in patients who had both emphysema and ILA. The impact of emphysema spatial capacity in mortality was higher in patients with ILA, compared with patients who had emphysema alone (p=0.04). Although previous mentioned studies indicate poor prognosis of ILA in COPD patients, a previous study showed that the presence of ILA was associated with a lower exacerbation rate in patients with COPD [84].
3. Prognosis of coronavirus disease 2019 in CPFE patients
Coronavirus disease 2019 (COVID-19), which is caused by severe acute respiratory syndrome coronavirus 2 virus, has been affected 170 million patients and caused more than 3.5 million death worldwide by June 2021 [91]. Among patients with COVID-19 disease, comorbidities such as chronic respiratory diseases, cardiovascular diseases, diabetes and cancer are reported to be associated with severe course of the disease [92-95]. Since there have been no data investigating prognosis of COVID-19 in CPFE patients, its prognosis may be assumed by reviewing data of each disease component. Accompanying ILD in COVID-19 was shown to be associated with higher mortality [96,97]. Presence of IPF rather than non-IPF ILD, old age, male sex, obesity, severe underlying ILD and low FVC were associated with poor survival in those patients [96,98]. In contrast, conflicting results have been reported regarding impact of COPD in COVID-19; some data shows that it is associated with risk of hospitalization and mortality [99,100], while some studies present no significant association of COPD in risk of hospitalization, intensive care unit admission and mechanical ventilation, and mortality [101-104].
Management
1. Non-pharmacologic treatments
1) Smoking cessations
The most important aspect during management of patients with CPFE is smoking cessation, because smoking is the most influential risk factor for both emphysema and IPF. Nearly all patients with CPFE have a history of smoking. Moreover, patients who continue to smoke show significant radiologic progression of CPFE lesions, compared with patients who quit smoking [77,105]. Patients who continue to smoke should be prescribed individualized counselling and drug therapy (e.g., nicotine replacement therapy, bupropion, or varenicline) [106]. Furthermore, because the presence of ILA is also associated with a history of smoking, smoking cessation should be recommended for patients with ILA to improve their prognoses [21].
2) Routine pulmonary function tests
Previous studies have reported exceptional lung function test outcomes in patients with CPFE, and therefore routine follow-up involving lung function tests should be performed in these patients because the results may reflect disease progression and prognosis. In patients with CPFE, lung function test results are typically regarded as normal or sub-normal, especially in terms of lung volume and FEV1/FVC. This is the result of counterbalancing opposite effects on lung compliance and elastance of interstitial fibrosis and COPD [24,77]. However, DLCO is disproportionately reduced because both processes cause alveolar capillary membrane disruption [24,107,108]. Longitudinal analysis of 5-year lung function data in patients with CPFE was performed by Kitaguchi et al. [109]. Notably, annual reduction of vital capacity and FVC were significantly greater in patients with CPFE than in patients with COPD; FEV1/FVC were improved in patients with CPFE during the followup period. However, DLCO and DLCO/vital capacity were more rapidly reduced in patients with CPFE than in patients with COPD. FEV1 is usually preserved in patients with CPFE because the traction effects of interstitial fibrosis prevent small airway collapse during expiration [12,110]. However, patients with CPFE involving rapid FEV1 decline had higher mortality [111]. Accordingly, assessment of DLCO and FEV1 in patients with CPFE may be important for monitoring disease progression and predicting patient prognosis.
In addition, the presence of ILA is associated with reduced lung volume in patients who smoke, which may result in pseudonormalization of FEV1/FVC in patients with COPD. Accordingly, these patients may exhibit preserved ratio impaired spirometry (PRISm) (FEV1/FVC ≥0.7 and FEV1 <80% predicted) according to spirometric assays [21]. Young et al. [112] performed 5-year longitudinal analysis of the COPDGene study and revealed that 36% of patients with PRISm progressed from GOLD 2 to GOLD 4. Accordingly, clinicians should perform routine lung function tests in patients with PRISm.
3) Pulmonary rehabilitations
There is increasing evidence regarding pulmonary rehabilitation in patients with chronic respiratory diseases [113]. In the GOLD 2021 guideline, pulmonary rehabilitation is recommended in patients with GOLD B-D COPD [1]. It has been shown to relieve respiratory and systemic symptoms, improve health-related quality of life and exercise tolerance, and reduce the hospitalization rate and mortality in patients with COPD [114]. Furthermore, positive outcomes have been reported in patients with ILD. In particular, pulmonary rehabilitation improves dyspnea, health-related quality of life, and exercise tolerance, although the magnitudes of benefits were lower than in patients with COPD [115-119]. Because two components of CPFE (COPD and ILD) are improved by pulmonary rehabilitation, this treatment may have favorable impacts on patients with CPFE. Tomioka et al. [120] performed a single-center retrospective study to compare the effects of 3-week pulmonary rehabilitation between patients with CPFE and patients with COPD. They measured lung function, exercise capacity using the 6-minute walking test, and health-related quality of life using the Short Form-36 (SF-36) questionnaire. Both groups showed lung function improvement, but 6-minute walking test results were only improved in patients with COPD. In the SF-36 test, patients with CPFE showed improved physical function (p=0.015) but worsened social functioning (p=0.044), whereas patients with COPD showed improvement in half of the subscales in the SF-36 questionnaire. There were significant differences in SF-36 score between patients with CPFE and patients with COPD in subscales of Δvitality (−6.3±22.4 in CPFE vs. 11.3±21.1 in COPD, p=0.009) and Δsocial functioning (−18.8±34.2 in CPFE vs. 5.3±35.9 in COPD, p=0.027). Although the CPFE group showed a reduced benefit of pulmonary rehabilitation, compared with the COPD group, this treatment remains helpful in terms of lung function and physical function in patients with CPFE. However, further prospective and multicenter studies are needed to confirm the positive effect of pulmonary rehabilitation in patients with CPFE.
2. Pharmacologic treatments
1) Inhalers
Many patients with CPFE use bronchodilator and inhaled corticosteroid (ICS) therapies in real-world clinical practice, but few clinical studies have assessed the efficacies of those drugs in patients with CPFE [66]. Dong et al. [121] conducted a prospective cohort study of patients with CPFE, which investigated clinical efficacy and safety profiles of ICS/long-acting beta-agonist (LABA) therapies by grouping patients into ICS/LABA users and non-users. At the 1-year follow-up, ICS/LABA users showed increases in FEV1%, FVC%, and DLCO% of 11.2%, 13.5%, and 12.8%, respectively. By contrast, non-users showed decreases of 14.2%, 16.8%, and 21.3%, respectively. Furthermore, radiologic findings also significantly improved in the ICS/LABA user group, whereas they worsened in the non-user group. Favorable outcomes have been reported in terms of acute exacerbation rates, but no significant differences were found in safety profiles. However, because of the non-randomized nature of the study and small number of enrolled patients, additional research is needed to confirm the benefits of this drug.
According to the 2014 French Practical guidelines, patients with IPF are recommended to use inhaled bronchodilator therapy in the event of airway obstruction [122]. However, for most patients with IPF, FEV1/FVC is >0.7 regardless of emphysema, due to the FVC-counterbalancing effect of lung fibrosis. In these patients, the effect of bronchodilator is not clinically significant. However, in patients with SAD measured through impulse oscillometry parameters, bronchodilator use showed improvement in symptoms, FEV1, and forced expiratory flow at 25%-75% of the pulmonary volume [123,124]. In the absence of SAD, bronchodilator therapy is ineffective. Furthermore, patients with emphysema should not use bronchodilator therapy, because the effect was not clinically significant in patients with emphysema alone. Because SAD may be an early phase of COPD, regardless of airway obstruction, bronchodilator use may be considered in patients with IPF who have SAD [125,126].
2) Antifibrotics
Antifibrotic drugs, including pirfenidone and nintedanib, are known to impede disease progression in patients with IPF [127-130]. Pooled data analysis of three global phase 3 trials of CAPACITY trials (studies 004 and 006) and the ASCEND trial investigated 1-year outcomes of pirfenidone in patients with IPF [128]. In this pooled analysis, the authors concluded that pirfenidone treatment reduced the FVC decline rate (-216 mL vs. -363 mL per year, p<0.001), enhanced progressionfree survival by 38%, and improved symptoms of dyspnea and 6-minute walking distance, compared with the placebo group. Although the study only enrolled patients with FEV1/FVC >0.70, the authors performed a subgroup analysis of FVC decline regarding FEV1/FVC of <0.80, 0.80 to <0.85, and ≥0.85. Similar results were achieved with nintedanib, an intracellular tyrosine kinase inhibitor, in patients with IPF. The INPULSIS trials (i.e., two replicate, 52-week, randomized, placebo-controlled, double-blind phase III trials) compared the efficacy and safety of nintedanib in patients with IPF [130]. Treatment with 150 mg of nintedanib twice daily in patients with IPF led to significant alleviation of FVC decline, compared with the placebo group (-114.7 mL vs. -239.9 mL per year, p<0.001). As in previous studies regarding pirfenidone, the INPULSIS study enrolled patients with FEV1/FVC >0.70. In subgroup analysis of INPULSIS study data, nintedanib alleviated FVC decline in both FEV1/FVC >0.8 and ≤0.8 subgroups [131]. Moreover, nintedanib alleviated FVC decline, regardless of emphysema presence in baseline high-resolution CT. These results imply that patients with IPF and airway obstruction or emphysema can benefit from both antifibrotic drugs. However, further studies are needed regarding patients with IPF who have FEV1/FVC <0.70 or different extents of emphysema according to quantitative assays.
3) Steroids and immunosuppressants
There have been no reports that proves efficacy of systemic steroids and immunosuppressants on CPFE patients. However, decision to use those drugs should be made regarding phenotype and disease status. As CTD-ILD have been shown to have potential beneficial effect with corticosteroids and immunosuppressant, those drugs should be considered to be applied in CTD-CPFE [132,133]. However, previous reports and guidelines presents limited role of systemic steroids or immunosupressants in COPD (emphysema) or IPF patients [1,134]. On the other hand, patients in exacerbation state of CPFE may need systemic steroids as it is both recommended in AECOPD and AE-IPF [1,134].
4) Other adjuvant therapy
Large proportions of patients with CPFE have been reported to exhibit dyspnea, and most of these patients require oxygen supplementation during a 5-year follow-up period [63,77]. In the advanced stages of chronic lung diseases such as CPFE, breathlessness is a considerable burden to patients and may not be sufficiently diminished despite comprehensive interventions [135,136]. Previous studies reported that the usage of morphine may be effective for relieving dyspnea symptoms, but other opioids have led to controversial outcomes [137-139]. Recently, several antidepressants have been studied for symptomatic relief in patients with chronic lung diseases. Mirtazapine, a noradrenergic and specific serotonergic antidepressant, was effective in treating chronic breathlessness in a multicenter, randomized, placebo-controlled, double-blind, feasibility trial [140]. Ninety-five percent of patients had either COPD or ILD, and all patients had a Modified Medical Research Council score ≥3 at baseline. After randomization, the mirtazapine group consistently showed a lower numerical rating scale score in weekly measurements over 1 month, compared with the placebo group. Other antidepressants, including sertraline and paroxetine, showed promising results for breathlessness relief in some small clinical studies, but further investigations are needed [141-144].
5) Vasodilators for pulmonary hypertensions
Pulmonary vasodilator therapy for pulmonary hypertension in patients with CPFE is not recommended, because there is minimal evidence to support this approach [145]. The usage of pulmonary vasodilators may worsen V/Q mismatch and hypoxemia [110]. However, a recent case report indicated that the use of tadalafil led to improvements in pulmonary arterial compliance and exercise capacity [146]. The benefits of pulmonary vasodilators in pulmonary hypertension should be investigated in future studies. Overall, pharmacologic treatments that may play a role in patients with CPFE are summarized in Table 2.
3. Surgical interventions
Limited roles of surgical interventions have been presented in patients with CPFE. A case report presented a successful lung volume reduction surgery in CPFE patients, which resulted in improvements in dyspnea, lung function and exercise tolerance [147]. In patients with severe disease, lung transplantation should be considered as an ultimate treatment. Takahashi et al. performed a single-center retrospective cohort study and reported clinical outcomes of lung transplantation in CPFE patients [148]. Five-year survival of CPFE patients were 79.0%, which was similar to IPF group, but more likely to suffer from primary graft dysfunction, acute cellular rejection and chronic lung allograft dysfunction.
Limitations
Our study is a narrative review that performed with unsystematic research, and thus may have limited value for making recommendations.
Conclusion
Concurrent interstitial fibrosis is often found in patients with COPD and patients with emphysema, because both diseases have strong relationships with cigarette smoking and old age. Patients with increased density in chest CT without a history of ILD are diagnosed with ILA. ILA is regarded as an early stage of ILD progression, and the presence of ILA has been associated with poor lung function, exercise capacity, healthrelated quality of life, and mortality in both patients with COPD and patients with emphysema. Furthermore, patients with diffuse interstitial fibrosis combined with upper emphysema are diagnosed with CPFE. The mortality of patients with CPFE is worse than the mortality of patients with emphysema alone, but results have been inconclusive when compared with patients who have IPF. Acute exacerbation, pulmonary hypertension, and lung cancer were most strongly associated with CPFE prognosis. Biomarker studies have suggested that the pathophysiology of CPFE is more closely associated with IPF, rather than COPD. For the management of patients with CPFE, smoking cessation is critical; routine lung function tests may be useful for monitoring disease progression. Pulmonary rehabilitation and drugs (e.g., ICS/LABA therapy, bronchodilator, anti-fibrotic agents, and antidepressants) have been recently proposed for use in disease control, but further investigations are needed. Surgical managements may have limited roles in severe disease.
Since the introduction of the disease entity called CPFE in 2005 by Cottin et al., numerous studies have been conducted to understand its clinical significance. There were several distinct features compared with ILD or emphysema alone especially in prognostic aspects. Therefore, clinicians should be aware of their clinical characteristics. However, most of the studies included small number of patients, and there were several discrepancies between study results. Further larger studies should be performed to fully understand its clinical implications. Also, there were little evidences on benefits of pharmacologic agents, which are needed to be studied in further studies. Patients with CPFE may be undertreated as pseudonormalization of lung function tests, who may be needed to be treated with COPD drugs including inhalers. In addition, as increasing performance of low dose chest CT, ILA have been more detected and recent evidences proved its clinical significance. Clinicians should be aware of these lesions as it may be associated with poor prognosis especially in emphysema or COPD patients.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Print
Print Download Citation
Download Citation