Subtype-Based Microbial Analysis in Non-small Cell Lung Cancer
Article information

Abstract
Background
The human lung serves as a niche for a unique and dynamic bacterial community related to the development and aggravation of multiple respiratory diseases. Therefore, identifying the microbiome status is crucial to maintaining the microecological balance and maximizing the therapeutic effect on lung diseases. Therefore, we investigated the histological type-based differences in the lung microbiomes of patients with lung cancer.
Methods
We performed 16S rRNA sequencing to evaluate the respiratory tract microbiome present in bronchoalveolar lavage fluid. Patients with non-small cell lung cancer were stratified based on two main subtypes of lung cancer: adenocarcinoma and squamous cell carcinoma (SqCC).
Results
Among the 84 patients analyzed, 64 (76.2%) had adenocarcinoma, and 20 (23.8%) had SqCC. The α- and β-diversities showed significant differences between the two groups (p=0.004 for Chao1, p=0.001 for Simpson index, and p=0.011 for PERMANOVA). Actinomyces graevenitzii was dominant in the SqCC group (linear discriminant analysis [LDA] score, 2.46); the populations of Haemophilus parainfluenza (LDA score, 4.08), Neisseria subflava (LDA score, 4.07), Porphyromonas endodontalis (LDA score, 3.88), and Fusobacterium nucleatum (LDA score, 3.72) were significantly higher in the adenocarcinoma group.
Conclusion
Microbiome diversity is crucial for maintaining homeostasis in the lung environment, and dysbiosis may be related to the development and prognosis of lung cancer. The mortality rate was high, and the microbiome was not diverse in SqCC. Further large-scale studies are required to investigate the role of the microbiome in the development of different lung cancer types.
Introduction
The human body, including the gut, skin, and other mucosal environments, is colonized by several microorganisms [1], and the interaction between these microbiomes and the immune system has been continuously studied. In healthy individuals, this interaction can contribute to immune homeostasis and susceptibility to infectious and inflammatory diseases [2].
The human lung serves as a niche for a unique and dynamic bacterial community related to the development of multiple respiratory diseases, such as chronic obstructive pulmonary disease [3], tuberculosis [4], idiopathic pulmonary fibrosis, and cystic fibrosis [5]. The lung microbiota is essential for barrier function, immune homeostasis, and anticancer immune surveillance via tumor antigenicity in healthy individuals [6]. Bacteria may disrupt the cell cycle by toxin production, resulting in cell growth with alterations in protein expression controlling DNA repair, cell division, and apoptosis [7], thus influencing the host immune response against malignant cells and promoting disease and cancer development [8]. These include Papillomaviridae, causing cervical cancer; Helicobacter pylori, contributing to non-cardia gastric cancer; and Schistosoma hematobium, responsible for bladder cancer [6].
Lung cancer is one of the most common cancers worldwide and is a leading cause of cancer-related morbidity [9]. Lung cancer is a complicated disease caused by interactions between host and environmental factors [10]. Among these risk factors, the microbiome plays a vital role in maintaining microecological balance and regulating host immune responses [11]. Recent studies using next-generation sequencing have revealed that the lung microbiome in patients with lung cancer differs from that in healthy individuals and that these microbiomes play a crucial role in immunity as well as cancer [12]. Therefore, understanding the mechanisms by which microbes present in the airways can influence lung cancer development and treatment could be beneficial to predicting cancer risk and improving treatment efficacy and safety [8].
Non-small cell lung carcinoma (NSCLC) is mainly classified into squamous cell carcinoma (SqCC), adenocarcinoma, and large cell carcinoma. The most common type of lung cancer is adenocarcinoma, which comprises approximately 40% to 60% of all lung cancer cases, whereas SqCC comprises 25% to 30% of all lung cancer cases [13]. Large cell (undifferentiated) carcinomas account for 5% to 10% of lung cancers and are relatively rare. Several genetic alterations, including activating mutations in epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK), c-ros oncogene 1 (ROS1), rat sarcoma (RAS), and v-raf murine sarcoma viral oncogene homolog B1 (BRAF), have been identified as drivers of tumorigenesis in NSCLC [14]. Although overall survival has improved owing to the use of targeted therapy for NSCLC patients with driver mutations and the introduction of immunotherapy, resistance eventually develops after using targeted anticancer agents for a certain period; additionally, targeted therapy for SqCC is limited [15]. Furthermore, only a small portion of patients with lung cancer achieve optimal and sustained efficacy from immunotherapy. There is no effective anticancer drug for patients who are resistant to immunotherapy, and the prognosis is poor [16]. Therefore, identifying the microbiome status is crucial to maintaining the microecological balance within the lungs, which could prevent the progression of lung cancer and maximize the effect of immunotherapy in NSCLC.
In this study, we aimed to investigate the microbial differences in patients with NSCLC according to the different subtypes of SqCC and adenocarcinoma, which are predominant in NSCLC.
Materials and Methods
1. Patient recruitment and sample collection
A total of 84 patients who were pathologically diagnosed with NSCLC were recruited from June 1, 2018, to June 31, 2020. Patients admitted for a lung cancer diagnosis in two tertiary hospitals—the Severance Hospital and Seoul National University Bundang Hospital, in South Korea. A bronchoscopy specialist collected bronchoalveolar lavage (BAL) fluid samples using a sterile bronchoscope.
2. Sample collection
Before bronchoscopy, all patients washed their mouths twice with a sterile saline solution. The patients were given topical anesthesia (lidocaine) using a nebulizer. Subsequently, patients were administered with midazolam and fentanyl, as recommended [17]. The bronchoscope was placed into the mouth of the patients and then placed into the lungs. BAL fluid was obtained according to a standardized protocol: when the bronchoscope arrived in the “involved” airway containing lung masses or lung nodules, the bronchi were flushed with 30 to 50 mL of sterile saline (0.9%). Approximately 15 mL of BAL fluid samples were obtained from each patient for sequencing analysis. BAL fluid samples were immediately placed at –70°C in a freezer, and DNA extraction was conducted within 24 hours.
3. DNA extraction, polymerase chain reaction amplification, and sequencing
According to the manufacturer’s instructions, total DNA was extracted using the Maxwell RSC PureFood GMO and Authentication Kit (Promega, Madison, WI, USA). We performed polymerase chain reaction (PCR) amplification using fusion primers which were targeting the V3–V4 regions of the 16S rRNA gene of the extracted DNA. The fusion primers 341F (5’-AATGATACGGCG ACCACCGAGATCTACAC-XXXXXXXXTCGTCGGCAGCG TC-AGATGTGTATAAGAGACAG-CCTACGGGNGGCWG CAG-3’) and 805R (5’-CAAGCAGAAGACGGCATACGA GAT-XXXXXXXXGTCTCGTGGGCTCGG-AGATGTGTATA AGAGACAG-GACTACHVGGGTATCTAATCC-3’) were used for bacterial amplification; the underlined sequence indicates the target region of the primer. We performed amplification under the following conditions: initial denaturation at 95°C for 3 minutes followed by 25 cycles of denaturation at 95°C for 30 seconds, primer annealing at 55°C for 30 seconds, extension at 72°C for 30 seconds, and a final elongation step at 72°C for 5 minutes.
The PCR product was identified by performing 1% agarose gel electrophoresis and visualized using the Gel Doc system (BioRad, Hercules, CA, USA). The amplified products were purified using a CleanPCR kit (CleanNA, Waddinxveen, Netherlands). The purified products at the same concentrations were pooled, and short fragments (non-target products) were cleared using CleanPCR. We assessed quality and product size with a Bioanalyzer 2100 (Agilent Technologies, Palo Alto, CA, USA) using a DNA 7500 chip. Following the manufacturer’s instructions, mixed amplicons were pooled, and sequencing was performed at CJ Bioscience, Inc. (Seoul, Korea) using an Illumina MiSeq Sequencing system (Illumina, San Diego, CA, USA). Detailed methods are described in the Supplementary Materials and Methods.
4. Statistical analysis
Categorical variables are reported as numbers (percentages). Continuous variables with normal distribution are reported as mean±standard deviation, while variables with abnormal distribution are reported as median with interquartile ranges (25th–75th percentiles). Depending on the normality of distribution, categorical variables were compared using the chi-square test, and continuous variables were compared using either an independent t-test or Mann-Whitney U test. p-values <0.05 were considered statistically significant. All statistical analyses were performed with IBM SPSS Statistics version 25.0 (IBM, Armonk, NY, USA).
5. Ethics approval and patient consent
The study protocol was approved by the Institutional Review Board of Severance Hospital, South Korea (IRB No. 4-0018-0313) and Seoul National University Bundang Hospital, South Korea (IRB No. B-1610/365-302). The study design was approved by the appropriate ethical review committee, and we obtained informed consents was from all participants.
Results
1. Patient characteristics
The patients were divided into two groups according to their pathologic subtype: adenocarcinoma and SqCC. The baseline characteristics of the two groups are listed in Table 1. The adenocarcinoma group had 64 patients (76.2%), and 20 (23.8%) belonged to the SqCC group. The mean age was 66.7±11.2 years, and the patients in the SqCC group were older (p=0.033). Male gender (51.6% vs. 95.0%) and smoking history (46.9% vs. 90.0%) were dominant in the SqCC group, and the mortality rate was significantly higher in the SqCC group (6.3% vs. 25%, p=0.035).

Demographics and clinical characteristics of patients
2. Taxonomy composition in patients with lung cancer based on pathological subtypes
Figure 1 depict the differences between lung microbiomes according to the pathological type. The dominant phyla in the adenocarcinoma group were Bacteroidetes (40.8%), Proteobacteria (24.9%), Firmicutes (24.1%), Fusobacteria (6.0%), and Actinobacteria (2.8%). In the SqCC group, Bacteroidetes (35.0%), Firmicutes (29.3%), Proteobacteria (27.8%), Fusobacteria (3.8%), and Actinobacteria (3.3%) were dominant.
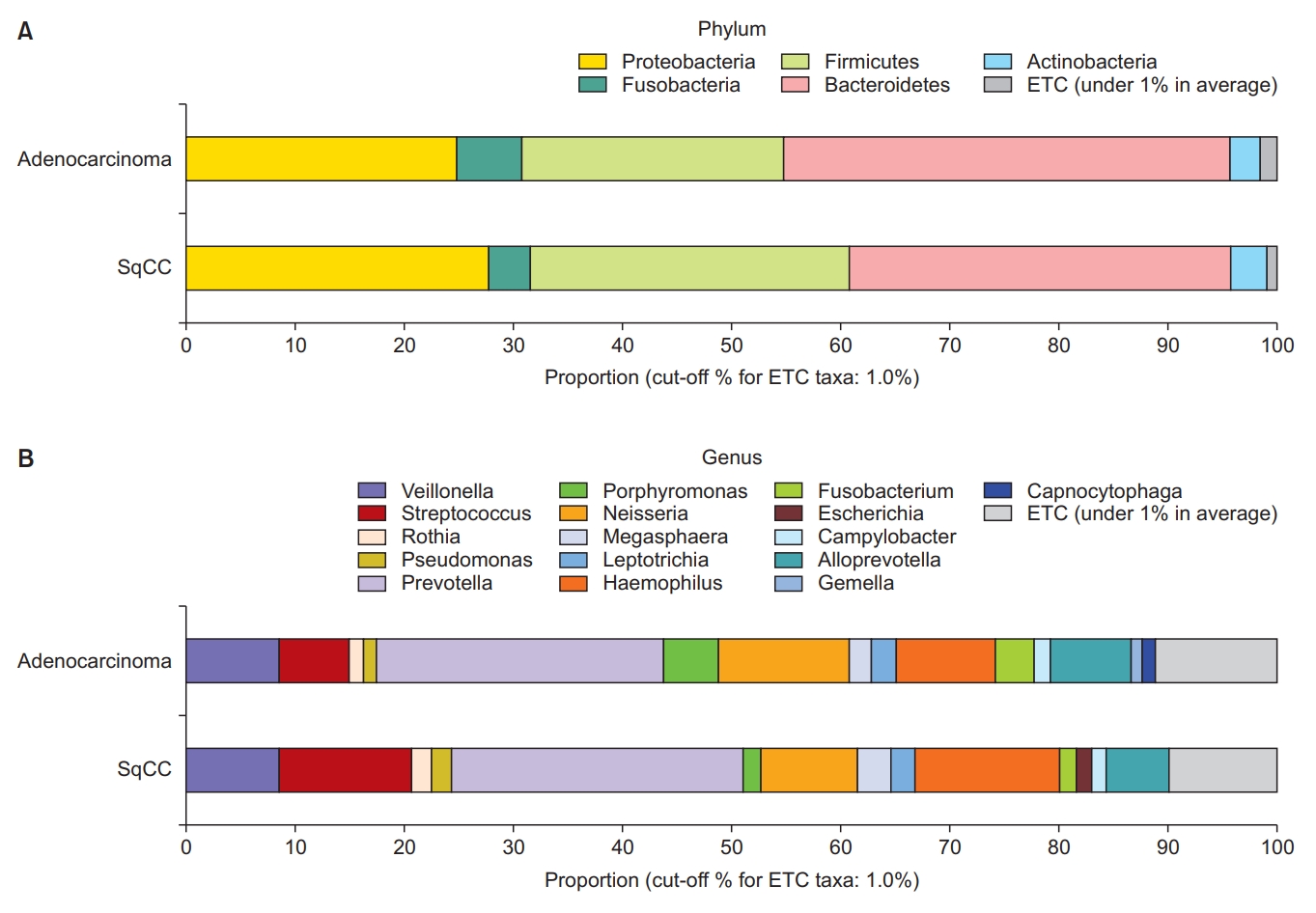
Taxonomic composition of the microbiome community between subgroups. (A) Dominant phyla based on the types of non-small cell lung carcinoma (NSCLC). (B) Dominant genera based on the types of NSCLC. SqCC: squamous cell carcinoma; ETC: et cetera.
The abundance of Fusobacteria differed between the adenocarcinoma and SqCC groups (Wilcoxon test, p=0.024) (Figure 2A), whereas that of Firmicutes, Bacteroidetes, and Proteobacteria were not statistically significant (Figure 2B–D). The abundance-based coverage estimator, Shannon, and Simpson indices were evaluated to estimate α-diversity in the lung microbiome, which summarizes the structure of an ecological community with respect to its richness (number of taxonomic groups), evenness (distribution of abundances of the groups), or both [18]. The operational taxonomic units of both groups exhibited statistically significant differences (p=0.006) (Figure 3A). Species richness differed between the two groups based on Chao1 (p=0.004) (Figure 3B), Simpson (p=0.001) (Figure 3C), and Shannon indices (p=0.0002) (Figure 3D).

Differential abundances of phyla. (A) Fusobacteria, (B) Firmicutes, (C) Bacteroidetes, and (D) Proteobacteria between the adenocarcinoma and squamous cell carcinoma (SqCC) groups.
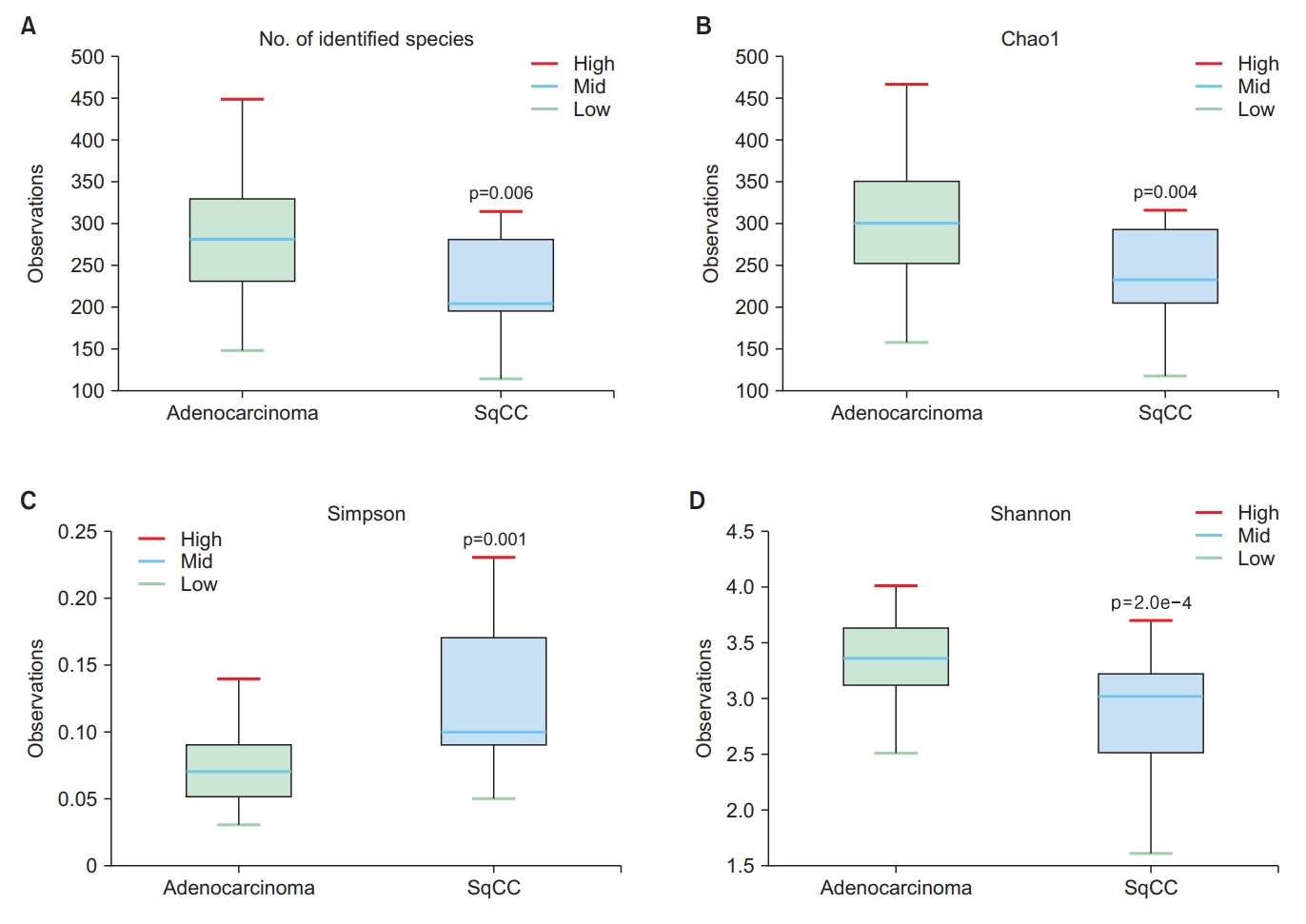
Comparison of the α-diversity in bronchoalveolar lavage (BAL) fluid microbiomes between the adenocarcinoma and squamous cell carcinoma (SqCC) groups. (A) Number of operational taxonomic units (OTUs), (B) Chao1 index, (C) Simpson index, and (D) Shannon index. Upper box, 2nd quartile; midline, median; lower box, 3rd quartile; whiskers, highest and lowest quartiles.
Principal coordinate analysis (PCoA) was performed to examine the similarity between the bacterial communities in each group. The Bray-Curtis distance was calculated to estimate the β-diversity in the lung taxonomy community structure in patients with NSCLC, which provides a measure of the degree to which samples differ from one another and can help elucidate the aspects of microbial ecology that are not apparent from the composition of individual samples [19]. A significant difference according to the pathological type was observed between the groups (p=0.011) (Figure 4).
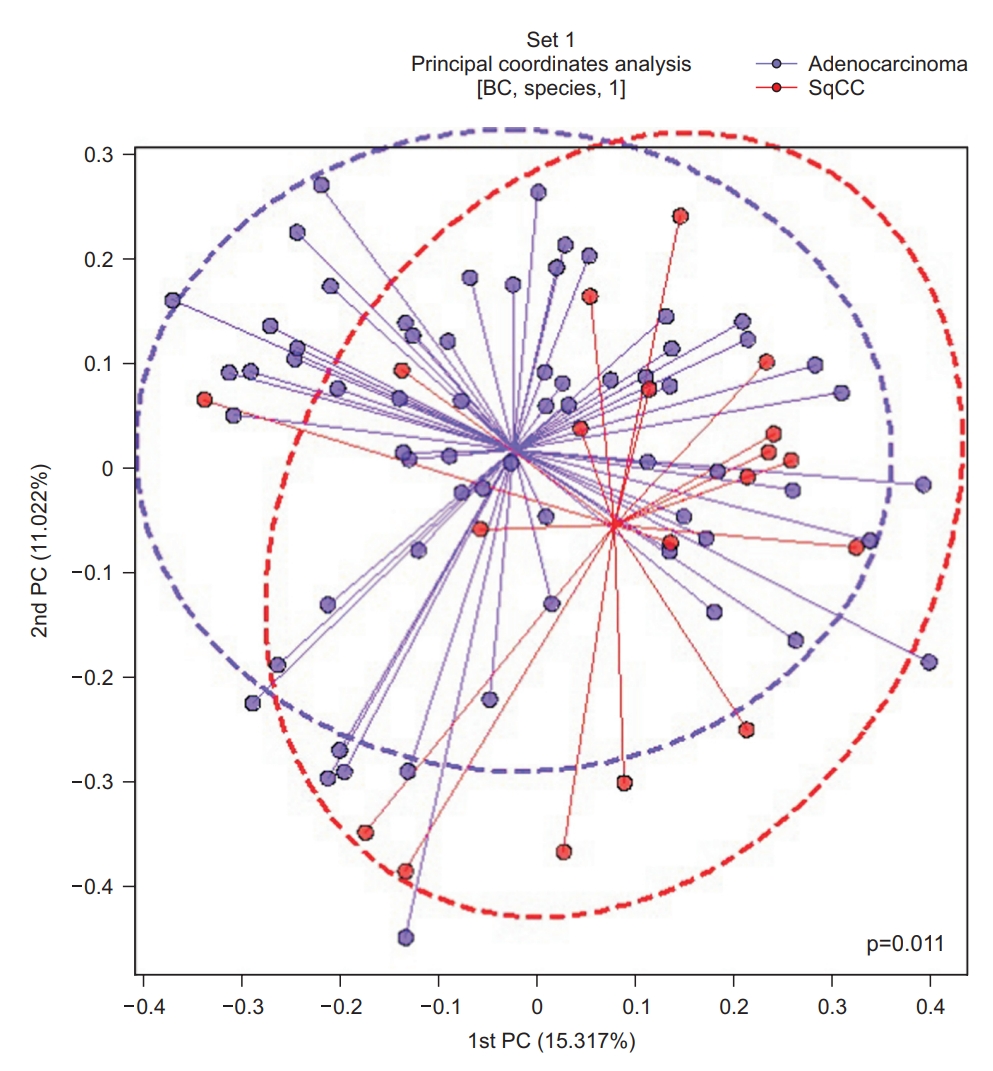
Principal coordinate analysis (PCoA) plot based on the Bray-Curtis (BC) distance of the BAL fluid microbiome between the adenocarcinoma and squamous cell carcinoma (SqCC) groups. PC: principal component.
We performed linear discriminant analysis (LDA) effect size (LEfSe) analysis to further evaluate the differences in these dominant genera between patients with NSCLC in the adenocarcinoma and SqCC groups, suggesting that the genus Actinomyces, which belongs to the phylum Actinobacteria, was significantly more abundant in the SqCC group (Wilcoxon test, p=0.049) (Figure 5). The genus with the greatest influence on the distinction between the two groups, with LDA score of 4.08, was Haemophilus, belonging to the Proteobacteria phylum (Figure 5).
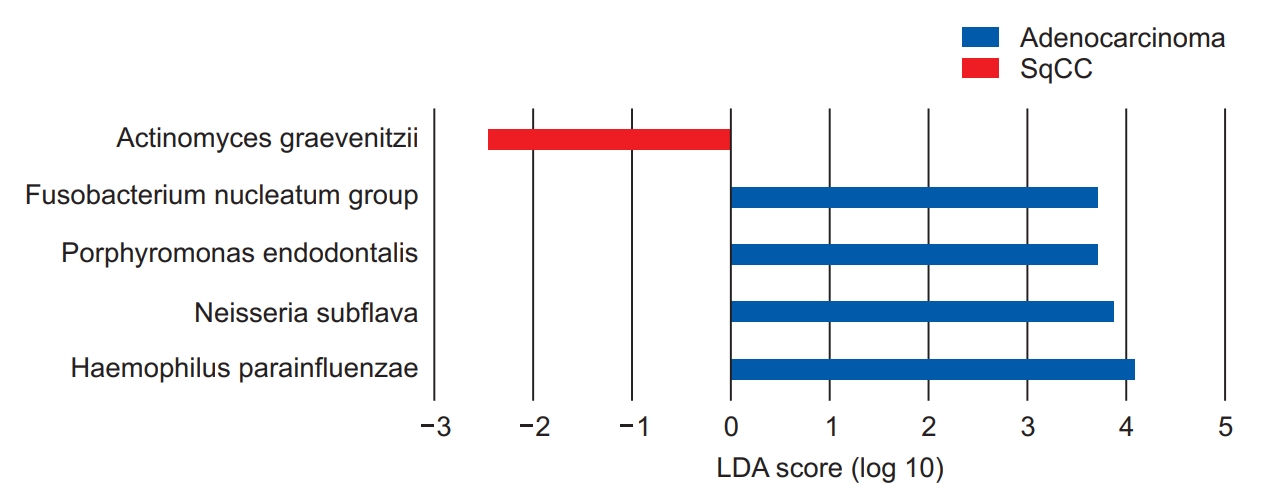
Linear discriminant analysis (LDA) effect size (LEfSe) analysis of the dominant genera between adenocarcinoma and squamous cell carcinoma (SqCC) groups. The blue and red bars indicate the taxa identified in greater relative abundance in patients with adenocarcinoma and SqCC, respectively.
Neisseria subflava, belonging to Proteobacteria, was dominant in the adenocarcinoma group with an LDA score of 4.07 (Wilcoxon test, p=0.029). These were followed by Porphyromonas and Fusobacterium with LDA scores of 3.88 (p<0.001) and 3.72 (p<0.001), respectively, in the adenocarcinoma group (Figure 5).
Discussion
In this study, we characterized lung cancer microbiota by analyzing BAL in patients with NSCLC from two hospitals using 16S rRNA sequencing. The diversity of microbiome was significantly reduced in SqCC comparing with adenocarcinoma.
Recent research has suggested strong associations between lung cancer and specific microorganisms through different mechanisms, including induction of host inflammatory pathways, production of bacterial toxins that alter host genomic stability, and release of cancer-promoting microbial metabolites [20]. For example, Veillonella is present in patients with lung cancer and plays a role in NSCLC pathogenesis [21]. It also plays a role in the increased infiltration of inflammatory cells (Th17 cells) and upregulation of the extracellular signal-regulated kinase/phosphoinositide 3-kinase (ERK/PI3K) pathway in bronchial epithelial cells [22]. Notably, PI3K pathway upregulation was previously shown to be an early pathogenic event in NSCLC, regulating cell proliferation, survival, differentiation, and invasion [23]. Concordantly, our study further revealed significant differences in the microbiome in NSCLC according to different pathologic types (adenocarcinoma and SqCC) exhibiting different prognoses.
Fusobacteria was a significantly abundant phylum in the adenocarcinoma group, and Proteobacteria was abundant in both groups; however, the abundance of certain species was different. Haemophilus parainfluenza, belonging to Proteobacteria, was the most dominant species in the adenocarcinoma group and is known to cause various invasive, chronic, and recurrent diseases [24]. A previous study revealed that the abundance of H. parainfluenza significantly differed between healthy individuals and patients with lung cancer [25]. We further found that it was more abundant in the adenocarcinoma group than in the SqCC group.
Porphyromonas endodontalis, belonging to Bacteroidetes, is known to be elevated in the sputum of patients with early lung cancer [26]. In our study, the proportion of early lung cancer (stage I/II) was higher in patients with adenocarcinoma (51.6% vs. 20.0%). Porphyromonas gingivalis, which belongs to the same phylum as P. endodontalis, promotes the survival and proliferation of epithelial cells by increasing PI3K/Akt signaling shortly after an infection, resulting in the inhibition of intrinsic apoptosis and increased expression of cancer stem cell markers CD44 and CD133 [27]. Although P. endodontalis has not yet been well studied, our study revealed its enhanced abundance in patients with adenocarcinoma, similar to P. gingivalis in oral cancer.
Neisseria species are associated with infections such as septic arthritis [28] and meningitis [29] among them, N. subflava is increased in patients with oral cavity cancer [30]. Fusobacterium nucleatum was also increased in oral cancers [31]. It promotes oral cavity cancer via direct interaction with oral epithelial cells through Toll-like receptors, increasing the expression of Toll-like receptor 2 (TLR2) in oral squamous cell carcinoma (OSCC) cells and interleukin 6 (IL-6) in cells and a mouse model [32]. F. nucleatum in esophageal cancer tissues has been associated with shorter survival, suggesting its potential role as a prognostic biomarker, which might also contribute to aggressive tumor behavior by activating chemokines, such as chemokine (C-C motif) ligand 20 [33]. The oral microbiome is correlated with the lung microbiome because the lungs are directly connected to the oral cavity [34]; thus, we can infer that N. subflava and F. nucleatum are associated with the development of lung cancer, especially adenocarcinoma. The microbiome was diverse in the adenocarcinoma group and similar to the microbiota of oral cavity cancer, suggesting that the oral microbial environment plays a crucial role in patients with genetic susceptibility to lung cancer development.
SqCC is associated with male smokers [35] and certain pathogens, such as that causing tuberculosis [36]. In patients with SqCC, Actinomyces graevenitzii was much more abundant than in the adenocarcinoma group. A. graevenitzii is a component of the oropharyngeal flora and is associated with pulmonary infections in some cases [37]; however, it has not yet been comprehensively studied. Our study revealed that it is related to SqCC.
Previous studies have reported differences in the clinical characteristics and treatment methods between the two subtypes [38]. Wang et al. [39] analyzed clinical characteristics of 48,296 patients with lung cancer by dividing them into adenocarcinoma and SqCC groups. The two groups differed significantly in many clinical characteristics, including age, sex, clinical/pathological stage, and treatment. Notably, they reported that the stage-specific 5-year overall survival rate differed significantly between the two groups even after propensity score matching, suggesting that the two cancers should be analyzed separately to provide a precise outcome. Similarly, our study identified significantly different microbiomes and PCoA plots depending on the cancer subtype. Furthermore, the microbiome was significantly more diverse in the adenocarcinoma group than that in the SqCC group. Also, there was significant difference of α-diversity between early (stage 1, 2 and 3a) and late stage (stage 3b and 4) of NSCLC (Supplementary Figure S1). As the microbiome plays a vital role in barrier function and immune surveillance, this low microbial diversity may also be related to the poor prognosis of SqCC.
Although diet and antibiotics usage, which might influence the microbial composition, were not investigated, a specialized bronchoscopist conducted sterile bronchoscopy, and we used 16S rRNA sequencing during the first diagnosis, which was prospective, informative, and sensitive compared with other conventional methods.
In conclusion, we identified that the microbial α- and β-diversities were different between the two types of lung cancer. H. parainfluenzae, N. subflava, P. endodontalis, and F. nucleatum were significantly abundant in the adenocarcinoma group, whereas A. graevenitzii was abundant in the SqCC group. Prolonged colonization and subsequent inflammation may disrupt the normal host immune barrier, eventually leading to cancer development. Further large-scale studies are required to determine the role of the microbiome in the development of different lung cancer types.
Notes
Authors’ Contributions
Conceptualization: Jang HJ, Cho YJ, Lee SH. Methodology: Jang HJ, Lee EK, Cho YJ, Lee SH. Formal analysis: Jang HJ, Lee EK, Cho YJ, Lee SH. Data curation: Jang HJ, Lee SH. Software: Lee SH. Validation: Jang HJ, Cho YJ, Lee SH. Investigation: Lee SH. Writing - original draft preparation: Jang HJ, Cho YJ, Lee SH. Writing - review and editing: Jang HJ, Lee EK, Cho YJ, Lee SH. Approval of final manuscript: all authors.
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Funding
This Study was Supported by a 2021-Grant from The Korean Academy of Tuberculosis and Respiratory Diseases.
Supplementary Material
Supplementary material can be found in the journal homepage (http://www.e-trd.org).
Comparison of the α-diversity in bronchoalveolar lavage (BAL) fluid microbiomes between early and late stage of non-small cell lung carcinoma.