What Single Cell RNA Sequencing Has Taught Us about Chronic Obstructive Pulmonary Disease
Article information

Abstract
Chronic obstructive pulmonary disease (COPD) affects close to 400 million people worldwide and is the 3rd leading cause of mortality. It is a heterogeneous disorder with multiple endophenotypes, each driven by specific molecular networks and processes. Therapeutic discovery in COPD has lagged behind other disease areas owing to a lack of understanding of its pathobiology and scarcity of biomarkers to guide therapies. Single cell RNA sequencing (scRNA-seq) is a powerful new tool to identify important cellular and molecular networks that play a crucial role in disease pathogenesis. This paper provides an overview of the scRNA-seq technology and its application in COPD and the lessons learned to date from scRNA-seq experiments in COPD.
Introduction
Although within a given individual the background genetic information (i.e., DNA) is the same regardless of the organ investigated, the cellular transcriptome varies dramatically between tissues and even between cells within the same tissue. Moreover, the transcriptome is dynamic; thus, even within a cell, the transcriptomic information may change day to day or even by hour to hour in response to environmental exposures, disease states or as part of the “natural” ageing process [1]. One of way of evaluating the dynamic cellular transcriptome is single cell RNA sequencing (scRNA-seq). scRNA-seq is a powerful new state-of-the-art technology that has revealed new insights on human biology and disease mechanisms by unraveling the mysteries of RNA transcripts within individual cells and elucidating different composition of cell types within tissues and organs [2]. In 2013, Nature Magazine chose scRNA-seq technology as its “method of the year” [3]. Although a detailed description of scRNA-seq is beyond the scope of this paper, briefly, scRNA-seq interrogates the nucleic acid sequence information within individual cells using leading-edge next-generation sequencing instruments. Compared with the traditional bulk RNA sequencing (bulk-seq) technology, which sequences RNA bases from a mix of millions of cells, scRNA-seq provides much higher resolution of RNA information, enabling the capture of RNA data from a single cell (rather than a mixed signal from millions of cells) and thus an improved understanding of composition of different cell types within tissues or organs [2]. Importantly, scRNA provides information on the cellular function in the local micro-milieu for each cell type, allowing investigators to better understand functional networks of cells and the impact of environmental exposures and disease processes on these pathways and networks.
Since its advent in 2009, there has been an exponential increase in the number of scRNA-seq papers in the literature from nearly zero in 2011 to more than 2,000 papers per year in 2020 [2]. This rapid escalation in the number of papers has been driven by a marked descent in the cost of scRNA-seq technology (now less than $1 US dollar [USD]/cell vs. >$1,000 USD/cell in 2011) [2] and the wide dissemination of software packages to analyze the complex sequencing datasets generated by the scRNA-seq experiments. As of 2021, there were more than 1,000 bioinformatics software tools available to analyze scRNA-seq data [4]. Although, similar to other fields of medicine, there has been a rapid increase in the number of publications of scRNA-seq technology in pulmonary disorders, it pales in comparison to the number of publications in cancer [5] or immunology [6] where tens of thousands of papers have already been published (compared with a couple of thousands in pulmonary medicine).
Main Uses of scRNA-seq in Biology and Medicine
Please see Table 1 for definition of terms used this paper. The lung tissue is composed of over 40 different cell types [7]. Given their complex interactions within the microenvironment of the lungs, it is impossible to identify individual specific cells types in a tissue sample using bulk-seq even with the latest deconvolution methods. Flow cytometry, while it can identify specific cell types at a single cell resolution, relies on a pre-determined set of protein markers (typically found on cell surfaces) and as such cannot be used to elucidate novel cell populations. Further, there is an absolute limit to the number of proteins that can be used to identify cell populations per each experiment, which reduces its ability to evaluate the entire population of cells within a tissue sample [8]. scRNA-seq, on the other hand, interrogates the entire genome without bias. Thus, it can reveal the entirety of the cell populations within a tissue sample, even without an a priori hypothesis. This approach can lead to the discovery of novel cell types. One prime example is the discovery of ionocytes in the airway mucosa. In 2018, two independent groups used scRNA-seq technology on human and mouse airway tissue and identified pulmonary ionocytes, which expressed the transcriptional factor, forkhead box I1 (FOXI1), and were the major source of transcripts for the cystic fibrosis transmembrane conductance regulator (CFTR) gene in the airways, and whose expression was muted in cystic fibrosis (CF) [9,10].

Frequently used terminology in single cell sequencing experiments
Another common use of scRNA-seq technology is to elucidate the cellular landscape of tissues such as airway mucosa or immune cells in the small airways in both health and disease. Traditionally, airway epithelial layer was thought to consist of ciliated pseudostratified epithelial cells, secretory cells, mucus glands, and basal cells in the large airways and ciliated cells, basal cells and club cells in the small airways [11]. With the advent of scRNA-seq, it is now accepted that in addition to these cell types, the airway mucosa also harbors ionocytes, tuft cells and neuroendocrine cells, which make up 0.08%, 0.01%, and 0.02% of the cell populations in the airway, respectively [12]. In the more distal airways, scRNA-seq experiments have revealed new cell types such as “hillock” cells, preterminal bronchiole secretory cells, and transitional club-alveolar type 2 cells (also known as alveolar type 0 cells) [12]. Although the exact functions of these novel cell types have not been fully elucidated, hillock cells may regulate cellular adhesion and squamous epithelial differentiation and thus may be involved in the pathogenesis of asthma [13]. Other rare cell types include brush cells, which may be involved in chemosensation and regulation of neurogenic inflammation in the airways [14] and tuft-like cells, which may be precursors to both brush cells and ionocytes [13].
Another application of scRNA-seq technology is to identify novel gene co-expression patterns, enabling the discovery of gene-regulatory networks within and across cell types. Gene-regulatory networks in healthy tissue provide insight on how cells grow and differentiate over “time” for each cell type and how these cell types interact with each other in situ. Using sophisticated bioinformatics tool, investigators can determine the trajectory of cell populations across a high-dimensional expression space from one starting point to multiple endpoints based on the complexity of the transcriptomic expression of cells. One form of this analysis is based on “pseudotime” [15]. A pseudotime in this context is a differentiation (or lineage) trajectory from the simplest (e.g., a pluripotent stem cell) to the most complex state (e.g., alveolar type 2 cells) based on their RNA signature. Thus, a pseudotime analysis provides an estimate of how cell types might dynamically evolve over time (e.g., from monocytes to macrophages) [15]. The scRNA-seq data can also be used to determine how transcriptomically “active” cells are in a given tissue. By quantifying the abundance of pre-mRNAs to that of mature spliced mRNAs for each gene, RNA velocity tools can provide an estimate of the cellular dynamics for each cell populations (i.e., how active these cells are) [15].
These gene-regulatory networks at a single cell resolution can be used to identify possible therapeutic targets and compounds. By comparing the scRNA-seq profile between diseased (e.g., chronic obstructive pulmonary disease [COPD] lungs) and normal tissue samples (e.g., healthy lungs), investigators can identify differentially expressed networks and pathways (for each cell type) as well as specific genes that may be perturbed in disease, from which potential therapeutic targets can be assessed [16]. scRNA-seq data are also becoming an integral component of functional genomics screening for possible therapeutic compounds. In these experiments, investigators perturb target cells in vitro and determine their response using high-throughput methods that involve scRNA-seq technology to generate data for assessment of cellular viability, proliferation and/or signaling [16]. scRNA-seq data can also be used to identify novel biomarkers or readouts for preclinical and early phase drug development and to assess for possible side effects of these compounds as well as for their potential efficacy [16].
Limitations of scRNA-seq
There are several notable limitations of scRNA-seq technology. First, although the costs of scRNA-seq technology has dramatically declined over the past decade, it is nevertheless still very expensive (approximately $0.80 USD per cell), which includes the cost of library preparation, cell capture, sequencing and quality control [17]. Since each experiment requires approximately 5,000 to 10,000 cells, the overall cost of scRNA-seq for each sample is between $4,000 USD and $5,000 USD. Additional consideration is the capture efficiency of cells. Because ambient RNA (from dead or broken cells) can increase the noise of the experiment and confound downstream analysis, it is essential that only single “living” cells be captured and sequenced. However, some samples may contain dead or dying cells in which case the processing step (e.g., cell sorting and capture) may introduce significant noise to the experiment. Moreover, high ambient DNA concentrations in a sample, which is caused by cell death or improper processing of samples, can cause “living” cells to stick together, forming doublets, triplets or more complex cell combinations, which cannot (or should not) be sequenced. Thus, these cell complexes must be dropped from subsequent analyses in order to maintain the integrity of the experiment. An acceptable capture rate is >70% [18]. However, some samples (e.g., bronchial brushes) may contain very fragile cells (e.g., epithelial cells), which may drop-out at a higher rate, threatening the integrity of the experiment [19]. Another disadvantage of scRNA-seq is its relatively low read depth (typically between 30,000 and 60,000 raw reads per cell). In comparison, bulk-seq experiments confer 5 million to 200 million reads per sample. Thus, scRNA-seq has trouble detecting small transcripts, leading to a non-uniformity of coverage of genes across the genome and transcript length bias [17]. A third limitation is the relatively high technical variation (i.e., noise) between experiments. The Human Lung Cell Atlas (HLCA), which published scRNA-seq data on 2.4 million cells from 486 individuals across 49 datasets, showed that the greatest variation across the dataset was related to technical issues including the “batch effect,” inclusion of low-quality samples, RNA degradation, and differences in sample preparation and RNA capture efficiency [18]. Batch effect is particularly egregious as scRNA-seq generally requires fresh tissue. Because tissue samples from patients (e.g., bronchoscopy samples) cannot be easily batched together, each sample is processed and sequenced on its own. This introduces significant noise to the experimental data across the samples, causing a “batch effect” [20]. Although there are statistical approaches to address batch effects, they may not fully mitigate the noise and may in certain cases eliminate some of the signal within the experimental data [20]. It should also be noted that during the handling of samples, some cells may experience an “artificial transcriptional stress responses” related to the stress of the experimental protocol including chemicals used to dissociate the cells and use of transport in media that lack specific nutrients or growth factors critical for maintaining the integrity of certain cell types [19]. Ambient temperature can also affect cell viability and transcriptome expression. For example, tissue dissociation at 37°C can alter cell transcriptome; whereas scRNA-seq experiments conducted at 4°C minimize these changes [21].
There is a unique limitation related to scRNA-seq experiments involving bronchial brush and to a lesser extent bronchoalveolar lavage (BAL) samples. Bronchial brush samples contain mostly epithelial cells (>70%) with a minor contribution by immune cells that are “trapped” in cilia or in the submucosal or intercellular layers. Epithelial cells are very fragile and when lifted out of their natural microenvironment, they quickly undergo apoptosis. When we applied standard protocols (e.g., 10 minutes digestion) on these samples, our overall capture rate was less than 15%. We were able to improve the capture rate to >70% by developing a novel protocol [19], which focused on: (1) immediate processing of bronchoscopy samples following their collection in PneumaCult-Ex culture medium (STEMCELL Technologies, Vancouver, BC, Canada); (2) use of Accutase (Innovative Cell Technologies, San Diego, CA, USA), an enzyme mixture with proteolytic and collagenolytic properties, rather than relying on the use of a non-specific protease such as trypsin; and (3) reducing the digestion time to 3 from 10 minutes. We have also published a BAL-specific protocol to optimize cell capture rate for scRNA-seq experiments [19].
General Workflows for scRNA-seq
Although there are many different workflows for creating a single cell suspension for scRNA-seq, there are some important commonalities. All protocols involve the following steps: (1) isolation of a single cell; (2) extraction and amplification of mRNA from each of the isolated cells; (3) library preparation for sequencing; (4) sequencing of the cell’s genetic material using a high-throughput next-generation sequencer; and (5) data analysis using a state-of-the-art bioinformatics tool [22] (Figure 1). Each step is technically challenging and creates an opportunity to introduce errors, which can be systematic or non-systematic. Generally, the impact of these errors becomes larger if the source of error occurs at the beginning of the workflow. There are many different scRNA-seq platforms [2]. Our lab in Vancouver uses the 10xGenomics platform, which creates microdroplets from a sample of single cell suspension. After creation of cDNA from RNA, the nucleic acid is amplified using polymerase chain reaction and sequenced from the 3’ end using Illumina Nextseq 2000 [23] (Illumina, San Diego, CA, USA).
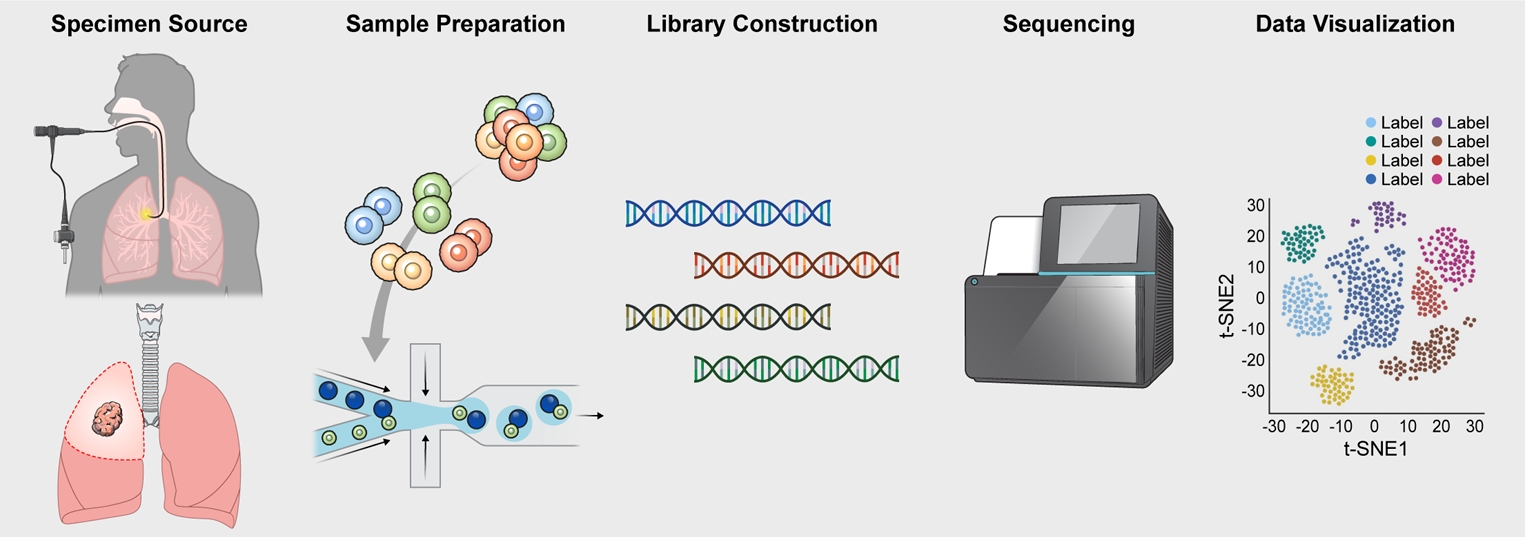
A typical workflow for a single cell RNA sequencing experiment. The single cell experiment starts with the procurement and processing of the target biospecimen. In pulmonary medicine, this is typically a resected lung tissue (from a lung cancer patient or explants) or bronchoscopic specimens (e.g., bronchial brush samples or bronchoalveolar lavage fluid). The cells from the retrieved tissue samples are then dissociated through enzymatic digestion; from which, a single cell suspension is created. The suspension is used to create cDNA libraries, which are then sequenced using a next-generation sequencer such as Illumina NextSeq 2000. The generated data undergo quality checks and pre-processing, after which, are subjected to dimensionality reduction for visualization and downstream analyses. The full details of this procedure are found in a publication by Gerayeli et al [19]. t-SNE: t-distributed stochastic neighbor embedding.
Data Analysis
Following sequencing, the generated data undergo several rounds of analyses (which collectively is called “pre-processing” of data). The primary analysis, which is typically performed on the sequencer, checks for the integrity of the base reads. If it passes through quality control checks, the data are then converted from BCL files to FASTQ files. Next, the reads in the FASTQ files are aligned with a reference genome, which can be done at the level of the gene or the transcript. This enables mapping of the reads to specific genes and allows for the quantification of gene expression levels [2].
Owing to a low capture efficiency, and shallow sequencing depth per cell, gene expression measurements obtained in scRNA-seq experiments are inherently noisy with many genes having a zero count. To enrich the signal-to-noise ratio of gene counts in scRNA-seq experiments, dimensionality reduction is essential [24]. This process involves the transformation of multidimensional, often noisy expression matrix, into a low-dimensional space (Figure 2). The most commonly used method is a principal component analysis followed by a uniform manifold approximation and projection mapping for data visualization, though other algorithms are also available including non-negative matrix factorization, and partial least squares and multidimensional scaling [24]. Following dimensionality reduction, scRNA-seq clustering methods are typically used for downstream analyses [25]. This step is then followed by biological annotation of data clusters, which can be done automatically using established algorithms, or manually using a curated set of reference markers [26]. To validate the labeling of the clusters, investigators often perform wet lab experiments including immunohistochemistry or flow cytometry.
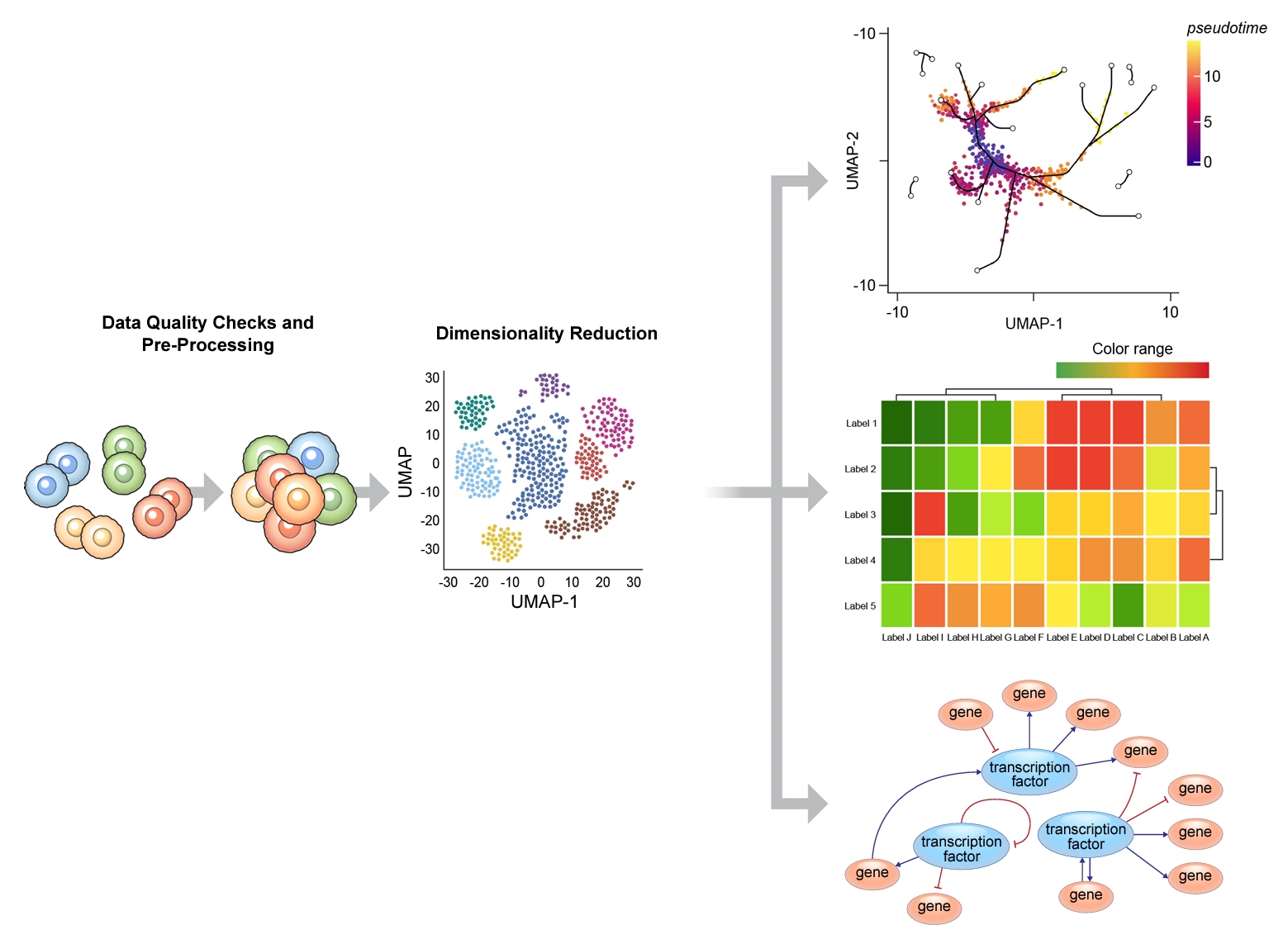
A typical bioinformatics pipeline to analyze data generated in a single cell RNA sequencing experiment. After quality checks and dimensionality reduction, bioinformatic tools are used to analyze the complex dataset to: determine differential gene expression at the level of single cell between diseased and normal states; map out the trajectory of cell lineage over time and between transitional states in disease and in health using pseudotime and/or RNA velocity analyses; and elucidate differential networks and pathways according to specific cell types in both disease and in health. t-SNE: t-distributed stochastic neighbor embedding; UMAP: uniform manifold approximation and projection.
Once the clusters are accurately annotated, differential gene expression comparisons can be made at the level of the clustered cell types between diseased and healthy individuals. To understand functional and/or biological significance of clustered cell populations, gene set enrichment analysis [27] or pathway analyses using Gene Ontology, Kyoto Encyclopedia of Genes and Genomes or Ingenuity Pathway Analysis databases are frequently performed. Additionally, investigators can run a pseudotime analysis to infer trajectory of cells at a single cell resolution [28] or a RNA velocity analysis to evaluate the dynamic state of cells in a sample in both health and disease (Figure 2) [29].
Applications in Chronic Obstructive Pulmonary Disease
COPD affects 385 million individuals around the world and is the 3rd leading cause of mortality. Aside from bronchodilators, there is a paucity of therapeutics to treat this disease [30]. The lack of disease-modifying therapeutics in COPD reflect its heterogeneity of endophenotypes and lack of good understanding of mechanisms that drive the disease [31]. scRNA-seq technology holds tremendous promise in addressing these gaps in knowledge. However, the application of scRNA-seq technology in pulmonary medicine is still at its infancy compared to other disciplines such as oncology. A quick PubMed search using search terms “COPD” and “scRNA-seq” reveals only 47 publications with nearly half being published in 2023. In contrast, there are 135 scRNA publications for pulmonary fibrosis and 633 studies for lung cancer in PubMed.
What have learned so far from these scRNA-seq studies in COPD? First, although the overall populations of major cell types in the airway mucosa are similar between patients with COPD and healthy individuals, on average, there is a reduction in the percentage of club cells [32,33] and an increase in the abundance of basal cells in the COPD epithelial layer [34]. Second, macrophages are the most abundant immune cells in the airways in both patients with COPD and healthy individuals [35]. However, in COPD airways, there are a lot more macrophages that are monocyte-derived, which are generally pro-inflammatory. In contrast, the airways of healthy individuals contain very few monocytes and monocyte-derived macrophages; most are tissue resident macrophages, which are anti-inflammatory and provide homeostatic function to the airways [35]. Moreover, macrophage/monocyte population of cells in COPD airways also demonstrate impairment in mitochondrial function, which may reflect altered cellular respiration and/or early senescence [36] and, there may be a subset of airway macrophages in the COPD but not in healthy airways that may be involved in ferroptosis [37].
Third, the genetic “COPD” hits from the genome wide association studies are enriched in a subset of type 2 pneumocytes expressing surfactant protein D and serine protease inhibitor A1 (SERPINA1) [38]. Fourth, there is enrichment of lymphocytes especially CD8+ cells and mast cells and general upregulation of the interferon-γ pathway in the small airways of patients with COPD [33]. Interestingly, compared with immune cells, or even epithelial cells, there are many more differentially expressed genes in the smooth muscle layer of the airways in COPD versus normal airways, which raises the possibility that airway smooth muscle cells may play an important role in COPD pathogenesis [12,38]. It should be noted that these scRNA-seq studies were performed on resected lung tissue samples, typically from patients undergoing lung cancer surgery or lung transplantation. As such, they may provide a biased view of disease severity, skewed towards those who are predisposed to oncogenesis or severe destructive disease. Notwithstanding this limitation, together, the current collection of scRNA-seq data in COPD indicates significant remodeling of small airways in COPD with a loss in anti-bacterial molecules, surfactants and other regulatory proteins that are crucial to host defenses and homeostasis in concert with the activation of pro-inflammatory, pro-apoptotic and senescent pathways.
Other Emerging Single Cell Omics Technologies
As highlighted previously, scRNA-seq suffers from important limitations such as tissue dissociation, which can not only make cells non-viable but also induce a transcriptional stress response even in the remaining “living” cells. Furthermore, scRNA-seq requires the use of fresh samples, which may be logistically difficult, expensive to procure and process and cause a significant “batch” effect between samples. One way to address these and other limitations is to use single nuclear sequencing or single nuclear RNA sequencing (snRNA-seq). Rather than sequencing single cells dissociated from tissue, snRNA-seq analyzes nuclei. snRNA-seq is possible even on frozen (and previously archived) samples and many different samples can be processed together and sequenced at the same time, reducing variations in data related to the “batch effect.” Altogether, snRNA-seq involves four crucial steps: (1) tissue processing; (2) nuclei isolation; (3) cell sorting; and (4) sequencing [39]. Because nuclear DNA is less abundant than mRNA, there may be significant challenges in enriching or depleting cell types of interest and important transcripts may be missed [39].
The assay for transposase-accessible chromatin using sequencing (ATAC-seq) is another single cell sequencing platform that enables the evaluation of the cellular chromatin landscape that may be perturbed by disease [40]. ATAC-seq uses a transposase, an enzyme that inserts a known DNA sequence to an “open” chromatin frame, to identify areas on the chromosome that are accessible for transcription. This technology has enabled nucleosome mapping, transcription factor binding, enhancer identification, and most importantly a better understanding of how epigenomic factors drive disease pathogenesis.
Summary and the Future
COPD is a highly complex disorder with multiple different endophenotypes. The advent of scRNA-seq and other single cell sequencing technologies is providing new insights on the heterogeneity of COPD and ultimately its disease mechanisms. Better understanding of disease pathways will enable discovery of novel therapeutic targets and biomarkers. To accelerate this process, it is crucial that single cell sequencing technologies be employed on tissue samples obtained from both ambulatory as well as surgical patients, and across the full spectrum of disease severity, who have been well-characterized clinically and phenotyped with imaging and outcomes data, along with patient symptoms. Moreover, larger and more ambitious studies are required in the future to fully realize the potential of single cell sequencing technology in elucidating disease pathogenesis and identifying novel therapeutic targets and biomarkers, which are urgently needed to better manage over 300 million patients worldwide, who are living with COPD.
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Funding
No funding to declare.