Identification of Serial DNA Methylation Changes in the Blood Samples of Patients with Lung Cancer
Article information

Abstract
Background
The development of lung cancer results from the interaction between genetic mutations and dynamic epigenetic alterations, although the exact mechanisms are not completely understood. Changes in DNA methylation may be a promising biomarker for early detection and prognosis of lung cancer. We evaluated the serial changes in genome-wide DNA methylation patterns in blood samples of lung cancer patients.
Methods
Blood samples were obtained for three consecutive years from three patients (2 years before, 1 year before, and after lung cancer detection) and from three control subjects (without lung cancer). We used the MethylationEPIC BeadChip method, which covers the 850,000 bp cytosine-phosphate-guanine (CpG) site, to conduct an epigenome-wide analysis. Significant differentially methylated regions (DMRs) were identified using p-values <0.05 in a correlation test identifying serial methylation changes and serial increase or decrease in β value above 0.1 for three consecutive years.
Results
We found three significant CpG sites with differentially methylated β values and 7,105 CpG sites with significant correlation from control patients without lung cancer. However, there were no significant DMRs. In contrast, we found 11 significant CpG sites with differentially methylated β values and 10,562 CpG sites with significant correlation from patients with lung cancer. There were two significant DMRs: cg21126229 (RNF212) and cg27098574 (BCAR1).
Conclusion
This study revealed DNA methylation changes that might be implicated in lung cancer development. The DNA methylation changes may be the possible candidate target regions for the early detection and prevention of lung cancer.
Introduction
Worldwide, lung cancer is the leading cause of cancer death1. Although the survival rate trends have increased in recent years (due to early detection by low-dose computed tomography [CT] screening and new therapeutic agents, such as molecular targeted agents or immunotherapy), the 5-year survival rate remains poor, at about 20%–30%23. Therefore, in the area of lung cancer research, it is an important task still to identify biomarkers that may be useful for early diagnosis.
The genetic changes found in lung cancer can mainly be explained by the interaction between permanent genetic mutations and dynamic epigenetic changes4. Since permanent genetic alterations in lung cancer have been studied extensively there has been substantial progress in molecular-targeted therapeutics in the treatment of patients with non-small-cell lung cancer (NSCLC)5. However, the overall survival rate reduction effect of the molecular targeted therapeutics for lung cancer is insignificant. Although somatic genetic mutations might play a major role in cancer development, epigenetic changes are known to play more variable roles, since they can either repress the expression of tumor suppressor genes or activate the expression of oncogenes46. Therefore, epigenetic changes could be a target to uncover biomarkers for the early detection of lung cancer.
Epigenetic changes are defined as heritable modifications in gene expression which do not alter the DNA coding sequence directly7. Over the past decade, they have been studied increasingly as biomarkers for the early detection of cancer and therapeutic targets89. Epigenetic changes observed in oncogenesis consist of aberrant DNA methylation patterns, histone modifications, and regulation by noncoding RNAs7. DNA methylation is the most studied epigenetic change and is known to repress gene expression and maintain genomic stability9. Therefore, DNA methylation study may give a potential clue for identifying biomarkers for early detection of lung cancer or to predict tumor progression.
We evaluated serial changes in genome-wide DNA methylation patterns in blood samples from lung cancer patients.
Materials and Methods
1. Study subjects and the preparation of tissue samples
For this study, we used blood samples obtained from a Korean chronic obstructive pulmonary disease (COPD) cohort10. They were prospectively recruited from 2012 to 2015 to observe clinical outcomes of Koreans living near cement plants. They will be followed up for 10 years and will be monitored for clinical information, blood/urine samples, and surveyed via questionnaire. The cohort recruited 445 patients from 2012 till December 2015. Among them, five patients developed lung cancer till the end of 2016, two of small cell lung cancer and three of NSCLC. Among them, blood samples were obtained for three consecutive years from three patients who had developed NSCLC (2 years before, 1 year before, and after lung cancer detection) and from three control subjects who did not have lung cancer and were matched for age and smoking history (Figure 1). Appropriate informed consent was obtained from all the patients, and the Institutional Review Board of Kangwon National University Hospital approved the study (IRB no. KNUH2012-06-007). All the COPD cohort study conduct adheres to Good Clinical Practice Guidelines and the tenets of the Declaration of Helsinki.
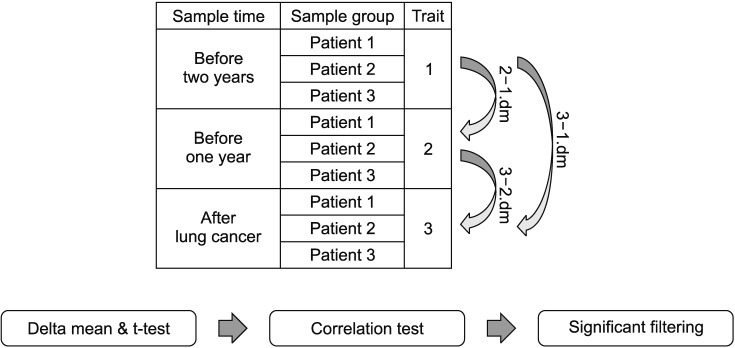
Blood samples were obtained for three consecutive years from three patients with non-small-cell lung cancer or control subjects and analyzed by delta mean and correlation test. Significant β changes were defined as when the delta mean of trait 2–1 and 3–2 was increased (≥0.1) or decreased (≤??.1), and significant correlation was defined when the p-value for the correlation test was >0.05. When both conditions were satisfied, significant differentially-methylated regions were obtained. dm: delta mean.
2. Genomic DNA preparation and DNA methylation profiling
We conducted an epigenome-wide analysis using a MethylationEPIC BeadChip kit, which covers the 850,000 bp cytosine-phosphate-guanine (CpG) site. The methylation value (β), a ratio between methylated probe intensity and total probe intensity, was interpreted as the proportion of methylation; β values range from 0 (unmethylated) to 1 (methylated).
The DNA quality was checked with a spectrophotometer (NanoDrop ND-1000 UV-vis; NanoDrop Technologies, Wilmington, DE, USA), and genomic DNA was diluted to 50 ng/µL using a Quant-iT PicoGreen quantitation assay (Invitrogen, Carlsbad, CA, USA). Bisulfite-conversion using an EZ DNA methylation kit (Zymo Research, Irvine, CA, USA) was carried out according to the manufacturer's protocols (Supplementary Methods).
3. Statistical analysis
We used methylation β values because they are more easily interpretable as methylation changes than M values are, the log2 ratio of methylated probe intensity and unmethylated probe intensity.
In three patients who had developed lung cancer, we assigned continuous labels to each patient in the order of the sample collection year (1, 2 years before lung cancer detection; 2, 1 year before lung cancer detection; and 3, after lung cancer detection). The mean differentially-methylated level (β value) of each trait and correlation test, according to continuous sample collection order, was evaluated. Significant β changes were defined when the delta mean of trait 2–1 and 3–2 was increased or decreased by a factor of at least 0.1. Significant correlation was defined as when the p-value for the correlation test was <0.05. Significant differentially methylated regions were obtained when both conditions were satisfied (Figure 1).
In addition, in the three control subjects, we assigned the continuous labels 1 to 3 in the order of sample collection year (1, first year; 2, second year; and 3, third year). The mean differentially methylated level (β values) of each trait and correlation test, according to continuous sample collection order, were evaluated. Significant β changes were defined when the delta mean of trait 2–1 and 3–2 was increased or decreased by a factor of at least 0.1, and significant correlation was defined when the p-value for the correlation test was >0.05. When both conditions were satisfied, significant differentially methylated regions were obtained.
All the methylation data processing, statistical analyses, and visualizations were conducted in R 3.0.2 (R Foundation for Statistical Computing, Vienna, Austria).
Results
The baseline characteristics of patients and control were summarized on Table 1.

Baseline characteristics of patients with lung cancer and matched control subjects
We identified 11 regions with significant methylation changes in the three patients with lung cancer. The 11 regions were as follows: cg02740582, cg05130642, cg07850982, cg16057201, cg16298462, cg19536810, cg19565594, cg21126229, cg25304620, cg25994476, and cg27098574. In addition, 10,562 regions were identified as having significant correlation for continuous years. Significant differentially-methylated regions satisfying both conditions were found in two regions: cg21126229 and cg27098574 (Figure 2). The related genes for cg21126229 and cg27098574 were RNF212 and BCAR1 . Figure 3 presents the clustering heatmaps.
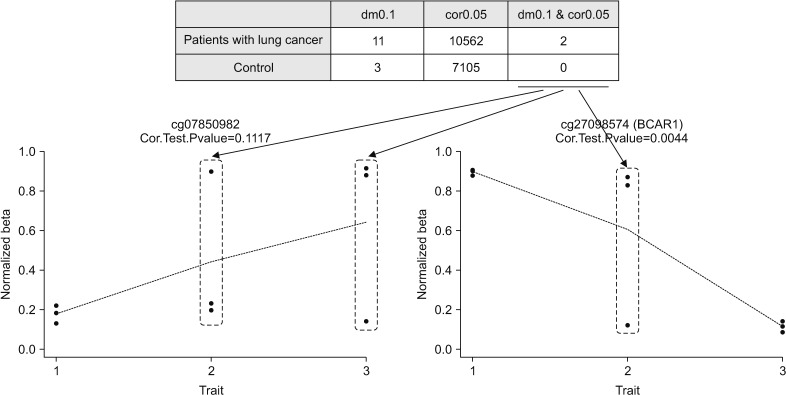
Significant differentially methylated regions with significant β changes and correlation. dm0.1, significant β changes of the delta mean with increased (≥0.1) or decreased (≤−0.1); cor0.05, significant correlation with p-value of >0.05.

Clustering heatmap of three patients with lung cancer. dm0.1, significant β changes of the delta mean with increased (≥0.1) or decreased (≤−0.1); cor0.05, significant correlation with p-value of >0.05.
Three regions with significant methylation changes were identified in the three control subjects. In addition, 7,105 regions were identified as having significant correlation for continuous years. However, there were no significant differentially-methylated regions which satisfied both conditions.
Meanwhile, to observe DNA methylation changes from another perspective, we analyzed cancer and control subjects by grouping them with same sample collection years. We sought to select a position that showed the difference between the cancer and the control samples at the same time, ultimately to show the difference between the cancer samples and the controls, and, at the same time, to show the position indicating the serial change. However, we did not find significant differentially-methylated regions with significant methylation changes and correlation for continuous years.
Discussion
In this study, we identified differentially methylated regions associated with the development of NSCLC in 3-year serial blood samples obtained from patients with and without NSCLC. Two genes, RNF212 and BCAR1 , that were associated with occurrence or early detection of NSCLC were found. Our study results may be used as a resource for finding biomarkers for the early detection of lung cancer.
Lung cancer is clinically manifested in various presentations and has few symptoms at the early stages. Early symptoms are subtle and non-specific and may lead to initial diagnosis being at an advanced stage for many patients with lung cancer. Although low dose CT screening was introduced as a promising strategy for the early detection of lung cancer1112, because it has several limitations, such as the possibility of lead time bias, a high rate of false-positive detection, and the as yet unanswered questions of radiation hazard effects, the debate continues13. The early detection of lung cancer may be more urgently needed. Epigenetic biomarkers such as DNA methylation and non-coding RNA may become alternative diagnostic methods to detect lung cancer in its earlier stages. The incidence of aberrant DNA methylation in cancer is higher than that of somatic mutation, and, since global DNA demethylation has been found in most lung cancers, it has the possibility to be a diagnostic marker for lung cancer49. Recent studies have suggested that gene-specific promoter DNA methylation may be a useful biomarker for the early detection of lung cancer or for the prediction of lung cancer outcomes49.
The aberrant promoter methylation changes in NSCLC have been identified in several genes from various samples. RASSF1A in sputum14, SHOX2 in bronchial aspirates15, and CDKN2A in bronchoalveolar lavage and exhaled breath condensates1617 have been found. In tumor tissues, Brock et al.18 showed that tumor recurrence was associated with differentially methylated regions of four gene promoters (p16, CDH13, APC, and RASSF1A); and Sandoval et al.19 identified hypermethylations of genes (HIST1H4F, PCDHGB6, NPBWR1, ALX1, and HOXA9 ) were associated with shorter survival. These studies have shown the possibility that DNA methylation biomarkers might improve diagnostic accessibility or prognosis prediction beyond standard staging. However, biomarkers for the early detection of lung cancer are in development only and it is expected that they are unlikely to be clinically applied within the next few years8. For a biomarker in clinical practice, it may be needed to be detected easily and early in blood. Promotor hypermethylation of six genes (RASSF1A, CDKN2A, RARb, CDH13, FHIT, and BLU) showed approximately 80% concordance between serum and tissue samples in NSCLC20. However, promoter methylation of CDKN2A, DAPK, PAX5b , and GATA5 in blood was much lower than in tissue samples21. These studies show that DNA promotor methylation in blood already has limitations for the early detection of lung cancer, because it assumes that DNA methylation changes in serum or plasma were found in DNA from apoptotic tumor cells, reflecting an advanced stage. Therefore, using an epigenome-wide associated study to identify new methylation changes of normal cells mediated by cancer cells, as immune cells, may open a new pathway for finding a biomarker for the early detection of lung cancer.
To understand DNA methylation changes induced by cancer, methylation across the whole genome should be measured simultaneously. However, whole-genome bisulphite sequencing may not be the most appropriate method because of its high cost and the technical expertise required to conduct it although it is considered a gold standard tool22. However, a microarray might be used as a popular and informative alternative. The Illumina Infinium BeadChips, the HumanMethylation27K and 450K BeadChip, have been introduced as tools for genome-wide DNA methylation profiling and have been affirmed as having easy and effective characteristics23. Recently, a more advanced tool containing over 850,000 probes, the Infinium MethylationEPIC (EPIC) BeadChip, with an increased genome coverage of regulatory regions, was introduced. With high reproducibility and reliability, the EPIC array is a significant improvement over the 27K and 450K microarrays24. Therefore, it may allow new valuable insights into the role of DNA methylation changes in cancer. This study, to our knowledge, is the first that uses the EPIC array to show DNA methylation changes in the blood samples of lung cancer patients.
Our study showed two genes associated with DNA methylation changes in lung cancer, RNF212 and BCRA1 . Studies have reported that the RNF212 gene is associated with variation in the genome-side recombination rate and acts to stabilize meiosis-specific recombination factors2526. There has been no reported association between RNF212 and lung cancer. Further research is needed. Meanwhile, BCRA is a wellknown gene whose role in lung cancer has been well studied. The BRCA gene acts as a tumor suppressor; its germ line mutation increases the risk of breast and ovarian cancer27. In early stage NSCLC, the DNA methylation of BRCA1 and high mRNA expression of BRCA1 is associated with poorer survival2829. However, to date, there is low-level evidence to support the use of BRCA as a clinically relevant biomarker in NSCLC. More precise and specific mechanism studies are needed.
This genome-wide DNA methylation study showed DNA methylation changes that might be implicated in the development of lung cancer. The DNA methylation changes may be candidate target regions for the early detection and prevention of lung cancer. Further investigation of these DNA methylation changes and related genes may enhance development of a biomarker and/or clinical applications for lung cancer.
Acknowledgments
This study was supported by a grant from the National R&D Program for Cancer Control, Ministry for Health and Welfare, Republic of Korea (1631210).
Notes
Author Contributions:
Conceptualization: Kim WJ, Hong Y.
Methodology: Moon DH, Kim WJ, Hong Y.
Formal analysis: Kwon SO, Kim WJ, Hong Y.
Data curation: Kwon SO.
Software: Kwon SO.
Validation: Kwon SO, Kim WJ, Hong Y.
Investigation: Moon DH, Kim WJ, Hong Y.
Writing - original draft preparation: Moon DH, Hong Y.
Writing - review and editing: all authors.
Approval of final manuscript: all authors.
The abstract of this paper was presented at the IASLC 18th World Conference on Lung Cancer, which was held 15–18 Oct 2017 at the Conference Center, Pacifico Yokohama, Japan.
Conflicts of Interest: No potential conflict of interest relevant to this article was reported.
References
Supplementary Material
Supplementary material can be found in the journal homepage (http://www.e-trd.org).
Supplementary Methods
Genomic DNA preparation and DNA methylation profiling.
