Introduction
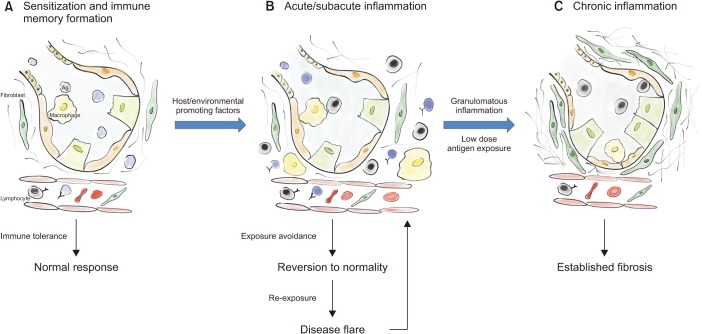
Hypersensitivity pneumonitis (HP), also known as extrinsic allergic alveolitis, is one of the most common interstitial lung diseases (ILDs)1,2. This particular ILD is characterized by exposure to an inhaled inciting antigen that leads to a host immunologic reaction determining interstitial inflammation and architectural distortion. The underlying pathogenetic mechanisms are still unclear, showing features of both type III and type IV hypersensitivity responses3,4. The sensitization process to the inciting antigen plays a pivotal role, turning a two-hit hypothesis in the most probable one (Figure 1).
HP epidemiology results to be challenging to determine considering the influence of many host, geographic and social factors5, also few cohort studies are reported in literature. But recently a one-year prevalence of 1.67-2.71 per 100,000 persons and a 1-year cumulative incidence of 1.28-1.94 per 100,000 persons were reported in a large American population6. With a 28% of 4-year mortality and a 52% of 7-year mortality fibrotic-HP6, the form with the worst prognosis, shows to have a better prognosis than idiopathic pulmonary fibrosis (IPF)7,8, but still a worse one when compared to some neoplastic conditions, such as thyroid, prostate and colorectal cancer9.
Furthermore, the absence of widely shared diagnostic guidelines and the lack of a “gold-standard” test for HP combined with the presence of several clinical and radiological overlapping features makes it particularly challenging to differentiate HP—mostly in its chronic and fibrotic form—from other ILDs, such as IPF10.
Therapeutic options for HP were limited to immunosuppressant drugs in the last decades, but recently the effect of nintedanib and pirfenidone has been tested with promising results in patients with chronic-HP (cHP).
This review aims to clarify the current state of the art of diagnosis and management of HP.
Diagnosis
1. Medical history and antigen exposure
HP seems to affect more often non-smoker elderly women6, even if epidemiologic data are contrasting, mostly regarding gender prevalence2,11,12,13. Several cases were reported also in children14 and among teenagers accustomed to vaping15. Cigarette smoke seems to have a double role in HP, it protects from developing the disease16, but if there is a positive smoking history it worsens the prognosis17. Several studies also reported a male predominance among HP subjects2,11,12,13.
Furthermore, other host factors may play a role in HP developing process. Just a minority of the subjects exposed to inciting antigens develop the disease, proofing that some sort of genetic predisposition is involved in the process18,19,20,21. Lopez and Salvaggio22 showed that just 5%-15% of the subjects with known exposure to inciting agents developed HP.
Another fundamental factor is the host work-related exposure. HP was firstly reported as Farmer's Lung in the early decades of the 20th century, making it clear that some class of workers are more prone to develop this disease. Among the others, we may find farmers, machinists, factory workers, and pigeon breeders23. Other environmental exposures, such as the possession of a pet-bird or an indoor hot-tub, significantly increase the likelihood of HP diagnosis. The main classes of inciting antigens are bacteria, fungus, mycobacteria, avianproteins, and chemical products.
HP symptomatology is aspecific and overlaps with several conditions, being characterized by shortness of breath, acute or slightly worsening, dry cough, malaise, fatigue, loss of appetite, chills, fever, etc.
Historically HP was classified into three categories acute, subacute and chronic; this classification was based both on symptoms behavior and disease course. While a recently proposed one, in order to solve some phenotype's overlap mostly between the subacute and the chronic form, promotes the presence of only two categories: the acute/inflammatory HP (aHP) and the chronic/fibrotic (cHP)24,25.
Influenza-like symptoms (i.e., fever, chills, cough, acute dyspnea, etc.) are more common in aHP; symptomatology usually begins a few hours following the antigen exposure and it may both last or increase over hours or days23,25. Similarly to what happens in working-related asthma, in aHP symptoms mostly decrease in presence and intensity after a suitable period of avoidance of the exposure and may proof, in this way, a causal link between expositions and symptomatology.
While cHP is characterized by a sneaky debut of symptoms characterized by slightly worsening dyspnea, dry chough, malaise, fatigue, loss of appetite23,25, that makes this form of the disease on a clinical standpoint very similar to other fibrotic ILDs and particularly similar to IPF. Repetitive and low-dose antigen exposure is the most common cause of cHP forms26.
The research of an inciting antigen may often be tricky: the percentage of cases with successful identification of the causative antigen stands below 40%27. Several methods were suggested in order to find it, such as questionnaires, precipitins, lymphocyte proliferation test and specific inhalation challenge (SIC), but none of those reached large consensus28. These methods present limited availability, lack of standardization and some of them may result in pretty expensive. There is also restricted amount of information about tests' characteristics, mostly regarding SIC.
2. Pulmonary function tests
Pulmonary function tests (PFTs)—as for every ILD—may reveal functional impairment and guide clinicians through patient management. Global spirometry and diffusing capacity of carbon monoxide (TLco) are useful non-invasive methods to determine disease course and loss of lung function. TLco and forced vital capacity (FVC) decline over time are predictive factors of survival in HP patients29,30. TLco particularly may predict exercise impairment being the most sensitive functional abnormality31,32. HP-related histologic changes, indeed, reduce gas exchange's area and influence pulmonary distension. A >10% FVC decline over 6-12 months showed to be a good predictor of worse prognosis33. However, when fibrosis is present, PFTs may be insensitive to detect small airway involvement34.
Besides the acknowledged utility, PFTs may not differentiate HP from the other ILDs37.
3. Radiology
Nowadays high resolution computed tomography (HRCT) plays a role of growing interest in HP diagnostic process and patient management. It is also recognized as the tool capable to identify HP findings with the highest sensitivity34.
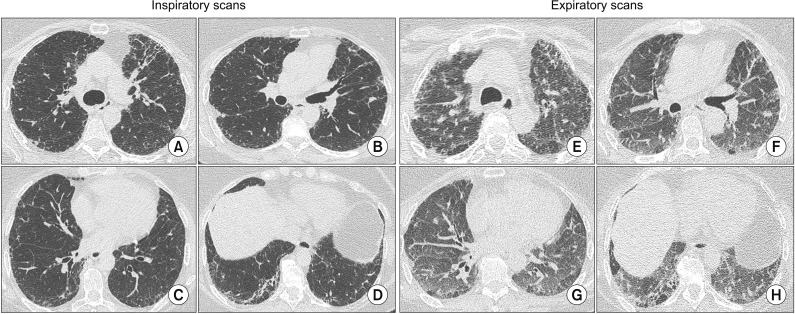
The main findings of aHP are multifocal and diffuse groundglass opacities (GGO), centrilobular GGO, signs of air trapping on expiratory phases studies and “headcheese sign.” This last sign is characterized by the contiguity of lobular areas with different attenuation levels (low, normal or high), which reflect heterogenous processes such as GGO, reticulation or fibrosis (high attenuation), normal parenchyma (normal attenuation) and air trapping (AT) (low attenuation)38.
The heterogeneity of HP, with both obstructive processes (small airways' obstruction) and infiltrative ones (interstitial phlogosis)38, is exemplified by the headcheese sign (Figure 2) and mosaic attenuation (MA). HRCT is useful to identify localized disease-related impairments and airway involvement. Among the ILDs HP is the one that more extensively exhibits MA, turning it into a prominent feature of HP39.
The main findings of cHP are architectural distortion, reticular opacities, peribronchovascular interstitial thickening, ill-defined centrilobular GGO, MA pattern, upper and middle zone predominance, sparing of basal lung areas and traction bronchiectasis, honey combing (HC) may be present (Figure 2)34. At its most advanced stages of disease cHP might show radiologic pattern hard to differentiate from either fibrotic nonspecific interstitial pneumonia (NSIP) or usual interstitial pneumonia (UIP) with HC. The differential diagnosis of HP with other ILDs might be tricky also because of these similarties40,41,42, mostly in cases where there is no identification of the inciting antigen.
Rarer are the cases in which cysts and emphysema are recognized on the chest computed tomography scans of HP patients, but they are still possible radiologic features of this heterogenous condition43,44.
Recently great attention was put on radiologic features of HP with different studies focusing on HRCT prognostic and diagnostic role. The great majority of the studies used a qualitative approach34, opening the door analysis' bias.
Salisbury et al.45 developed a radiologic predictive model for HP diagnosis. Their analysis enlightened that when a major extension of AT and MA compared to that of reticulation is present, combined to a diffuse axial distribution, there is less than 10% chances to make a false-positive diagnosis of HP45. Silva et al.41 explored the role of HRCT in solving the challenge of differential diagnosis among HP, IPF, and other ILDs. A confident differentiation between these different entities was possible in nearly half of the cases, underlying once again how tricky it may be relying just on radiologic findings for differential diagnosis. Barnett et al.42 recently focused their attention on MA and its role in the differential diagnosis with IPF. The presence of the headcheese sign resulted a more confident feature to lead to differentiation between cHP and IPF than just the extension of MA, challenging the faith of the scientific community42,46.
Some studies were performed in order to highlight the prognostic role of radiologic features in HP. Chung and coworkers showed how the presence of AT and MA in cHP subject may positively influence the survival47. Salisbury et al.48 successfully identified three unique radiologic groups of HP patients with different prognostic outcomes. The presence of HC in cHP determined a prognosis superimposable to the IPF one, worse than the prognosis of patients with just cHP; as expectable, patients without fibrotic changes were the group characterized by the better prognosis48. This radiologic phenotypes seems easy to adopt and useful in stratifying HP patients49.
The quantitative analysis through the CALIPER software was the fundament of Jacob and coworkers' study50. This is one of the few studies relying on quantitative analysis methods in HP. The main findings of this work were that the automated analysis performed better than visual scores and was able to stratify HP population based on pulmonary vessel volume (PVV). Patients with cHP and a high PVV presented a prognosis similar to IPF patients50.
4. Bronchioalveolar lavage
The use of the bronchoalveolar lavage (BAL) technique is pretty common in the ILD field, even more after the release of the last guidelines for IPF diagnosis51. American Thoracic Society (ATS) already addressed this issue in 2012, when an official guideline about the clinical utility of BAL fluid (BALF) analysis in ILD was released52. BAL is often performed in HP patients both in the acute and in the chronic forms. Several studies reported data about BALF cellularity in these conditions30. By the way, it is still unclear the role of BAL both in HP diagnostic process and in HP patients' management.
Lymphocytosis is, doubtless, the BALF parameter more often taken into account in HP studies and diagnostic predictive models. A higher percentage of lymphocyte in BAL may be a positive predictor of HP diagnosis, even if a BALF lymphocytic profile may be found also in sarcoidosis, NSIP, and organizing pneumonia52. Higher levels of lymphocytosis (>30%-40%) seem to better correlate with aHP. While cHP may show a slightly increased lymphocytic count (>20%) or even a normal one, mostly in patients with radiologic UIP53,54. Some authors argue that a higher lymphocytic count may be a positive predictive factor for survival, considering those forms of HP acute ones and more prone to disease resolution55 and response to corticosteroid treatment56. On the other hand, a BAL lymphocytosis >30% seems to be a useful tool in the tricky differential diagnosis between cHP and IPF, making IPF diagnosis very unlikely in such a scenario57.
Besides all the efforts widely shared thresholds for BAL lymphocytosis have not been identified yet25,28.
Also the study of T cells population was carried on HP patients' BALF, but not definitive findings supporting its use in daily clinical practice have been made25,52. Patients with aHP seem to have a higher percentage of CD8+, which lowers after exposure avoidance, at the same time patients with cHP seem to have a CD4+ predominance58,59. When talking about the CD4/CD8 ratio, a nonspecific and insensitive trend towards lower levels seems to characterize HP25,52,54.
5. Histology
The integration of clinical, radiologic and pathologic findings remains the gold standard for the diagnosis of every subtype of ILD, but recently we are moving toward a world where less invasive procedures are performed on patients. Constantly growing importance is given to radiologic features, that in several cases may guide clinicians to a confident diagnosis even in the absence of histopathologic specimens.
Surgical lung biopsy (SLB), as for the other ILDs, is the gold standard for tissue sampling60, besides complications related to this invasive procedure. Other systems like transbronchial biopsies and transbronchial criobiopsies have been proposed as alternatives, but no formal validation and/or international consensus was reached about their use in the diagnostic process of ILDs51,61,62.
Being HP a condition with heterogenous histologic alterations SLB providing a greater amount of tissue should probably be the elective tissue sampling procedure in order to have more chances to find the most characteristic lesions.
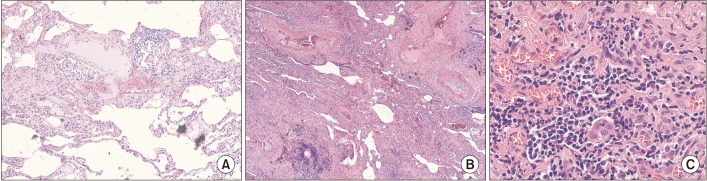
The classic histologic triad of HP is composed by peribronchiolar diffuse interstitial inflammatory infiltrates, chronic bronchiolitis and peribronchiolar giant cells (with or without non-caseous and non-necrotic granulomas) (Figure 3)63,64,65. In the latest stages of disease also UIP may be found.
Histology also plays a role in the prognostic stratification of HP subjects. The UIP pattern, when found, correlates with a worse prognosis29. Also airway centered fibrosis and fibrotic NSIP show a worse prognosis when compared to peribronchiolar inflammation with poorly formed granulomas and cellular NSIP66.
6. Actual diagnostic models and lack of consensus
Some predictive and diagnostic models were recently proposed to facilitate clinicians in the hard task represented by making a confident HP diagnosis24,25,28,37,67. None of them reached a wide consensus, but at least they aroused effervescent discussions about this topic68,69,70,71. And nowadays both the ATS and the American College of Chest Physicians (ACCP) are promoting initiatives aimed to draft clinical evidence-based guidelines for HP46,70.
Walsh and coworkers perfectly painted how challenging it may be reaching consensus about HP diagnosis72. In their study the inter-multidisciplinary team (MDT) agreement for HP diagnosis was incredibly poor, making it clear to everyone that even clinicians with a proven expertise in the ILD field may find it difficult to make HP diagnoses that meet the consensus of the other groups.
Furthermore, different groups of experts are also proposing different classifications of the disease making the HP field even more chaotic. We are all used to the historical classification with the three forms of HP: acute, subacute and chronic. That was recently overtaken by a more simplistic and binary one: aHP and cHP. This one, thus appearing too simple, seems to be useful30 and also supported by recent radiologic findings48,49.
One of the key points of HP diagnosis, in patients with compatible respiratory symptoms, is the recognition of the inciting antigen. All the proposed diagnostic models agreed on that. A careful clinical history should be collected and it is fundamental for HP diagnosis, nevertheless in several cases the inciting antigen may remain elusive27. The IPF guidelines and Fleischner Society's White Paper also suggest as one of the very first steps of the diagnostic process to check for possible exposures in patients suspected to have IPF, being HP a possible alternative diagnosis51,73. Vasakova et al.25 developed an exhaustive questionnaire, but it is still lacking of formal validation. The same group of authors proposed the definition of “cryptogenic HP” for all those cases of truly unknown origin, but this terminology was strongly criticized70,74.
All the available diagnostic models also agree about the absence of a “gold standard” test for HP diagnosis. However, radiology is nowadays having a pivotal role in the diagnostic algorithm of ILDs and HP is not excluded. Radiologic findings may indeed guide the diagnostic process towards other steps, such as what happens in IPF24. Radiologic features like extensive MA were typically thought to be expression of HP: Fleischner Society's White Paper reported “extensive” MA as most consistent with cHP than IPF73, while the last international guidelines indicate that “marked” MA should arise the doubt of IPF alternative diagnosis51. However recently we witnessed an attempt to change of perspectives when the headcheese sign was proposed as specific for cHP and inconsistent with IPF42. The more relevant role undertaken by HRCT leads to a minor rate of invasive procedures' execution, similarly to what happens with IPF tissue sampling is always less common such as BAL. These procedures are often relegated to the more challenging cases when, lacking the inciting antigen's identification and typical radiologic pattern, the diagnosis' confidence is low.
As previously reported, lymphocytosis is a characteristic finding in HP patients, nevertheless no shared threshold for it has been defined yet. Morisset and coworkers in their modified DELPHI survey reported consensus among international experts for high confidence HP diagnosis without the execution of lung biopsy in the right clinical contest (recognized exposure and HRCT consistent with cHP) when BAL lymphocytosis was >40%28. While Salisbury et al.24 proposed a 20%-30% threshold in their diagnostic model, which is mostly focused on cHPs.
The integration of the information conveyed by clinical history, lab tests, PFTs, HRCT, BAL analysis, and histology should be processed and discussed by MDTs in order to decide the best diagnostic strategy and reach a confident diagnosis. The importance of MDTs is growing constantly in the ILD field and the evidence-based algorithm proposed by Salisbury et al.24 takes the move from an MDT evaluation and wisely proposes a pathway that aims to reach the highest diagnostic confidence using the less amount of invasive procedures possible.
Treatment
1. Antigen avoidance and immunosuppressants
Guidelines for HP treatment are not available at the moment. All the therapeutic regimens proposed to derive mostly from observational studies and very few randomized trials.
The recognition of the inciting antigen is useful not only for diagnostic purposes but also for patients' management. HP is recognized as a condition driven by a massive autoimmune response to inhaled antigens, then the first step of HP treatment is the antigens' avoidance, when possible. This is mostly effective in the early/acute phases of disease and less effective in the chronic forms of HP, when fibrotic changes intervene27,69,75,76. Furthermore, in cHP fibrosis may progress even if the subjects are not exposed to the antigens anymore.
Another cornerstone of HP treatment is immunosuppressant treatment, for the same reason.
Corticosteroid treatment in HP is supported by weak evidence. There is just one randomized trial and it is short and small77. The use of predinosolone tapered along 8 weeks against placebo was evaluated in cohort of 36 patients affected by Farmer's Lung. All the patients avoided contact with farms for the duration of the study. No differences were found in pulmonary function parameters, besides a significant difference in TLco. There was also no effect of corticosteroid treatment on mortality. Corticosteroids seem to be more effective on aHP when the phlogistic process is more abundant. Some other non-randomized trials and observational studies supported these findings75,76. De Sadeleer et al.76, in their retrospective study, showed that the steroid treatment had no benefit on cHP patients, while it was effective on lung function and mortality of aHP patients.
Other immunosuppressant therapies have been studied in HP patients, such as mycophenolate mofetil (MMF) and azathioprine (AZA)78,79. Both treatment regimens seemed to be effective improving TLco in cHP patients in the study of Morisset et al.78, not showing any benefit on survival. The study conducted by Adegunsoye et al.79, instead, did not show any effect of both AZA and MMF on lung function. Nevertheless, cHP patients who needed to undergo immunosuppressant therapies showed a worse survival rate. However, an early transition to steroid-sparing drugs seems to reduce the incidence of treatment-emergent adverse events79. Rituximab is also sometimes used off-label as salvage treatment in refractory forms of cHP80.
Inhaled corticosteroid therapy is used on anecdotal basis81.
2. Antifibrotics and future perspectives
The prototype of the progressive fibrosing phenotype ILD is IPF, but this phenotype also develops in cHP. No matter what the lung injury causing agent is, fibrotic ILDs show similarities in disease presentation, behavior and also in the pathogenic process underlying the fibrotic process (Figure 1), which usually drives to irreversible loss of epithelial or endothelial barrier integrity, destruction of the lung architecture and loss of lung function82. Some authors took the moves from this assumption to test effectiveness of anti-fibrotic drugs in this cluster of diseases (Table 1).
Recently the results of phase III randomized placebo-controlled clinical trial—the INBUILD study (NCT02999178)—were released and opened the doors to the safe use of nintedanib, an intracellular tyrosine kinase inhibitor, for fibrosing ILDs83. Flaherty et al.83 showed that nintedanib was effective in reducing the annual rate of FVC decline when compared to placebo in a cohort of 663 subjects. Patients with cHP represented 26% of the enrolled population, turning this a fundamental study for the management of this condition.
An open-label proof of concept study (NCT02496182) explored the efficacy of pirfenidone as add-on therapy to immunosuppressant in cHP84. Besides not showing improvement of lung function test parameters, there was a significant St. George Respiratory Questionnaire's total score improvement in the pirfenidone group. Addition of pirfenidone seems to have a tolerable safety profile and promising results in cHP patients.
Some other randomized clinical trials (RCT) exploring the effects of pirfenidone on cHP are on their way. A phase II placebo-controlled randomized controlled trial—the RELIEF study (EudraCT 2014-000861-32 DRKS00009822)—will soon explore the efficacy of pirfenidone in fibrosing ILDs and cHP is recognized as one of the inclusion criteria. Another phase II study (NCT02958917) is going to explore the effect of full dose pirfenidone in cHP patients85.
Lung transplant may be a resolutive treatment in cHP at its last stages of disease, such as in other ILDs.
Conclusion
HP remains a challenging condition to diagnose and manage because of its heterogenous clinical, radiologic, histologic and functional features. Lots of efforts are put in making order in the ILD field, but HP remains still a foggy region—even if it is the third most common ILD.
Even if in the last decade the interest around HP was constantly growing, neither internationally accepted classification nor guidelines based on international consensus for both diagnosis and clinical management are available for this disease. Several classification proposals and diagnostic models were advanced by different groups but still have not received any prospective validation. The scientific community is anxiously waiting for the guidelines supported by the ATS and ACCP that are on the line and would facilitate the design of the next clinical studies.
Diagnosis is particularly tricky in the latest stages of cHP when it may be confounded with IPF also in expert contests. MDT meetings play a pivotal role both in the diagnostic process and in the decision making and patient management.
Immunosuppressant therapy, which—together with antigen avoidance—is effective on aHP, is now flanked by the evidence of the effectiveness of nintedanib in reducing the rate of lung function decline in cHP. Other studies about pirfenidone use in HP are ongoing.
However, several questions about this condition remain still unanswered and there is plenty of room for more studies to come and focus their attention on different aspects and pitfalls of HP.
 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Print
Print Download Citation
Download Citation