Introduction
Chronic obstructive pulmonary disease (COPD) is the fourth leading cause of death worldwide, with cigarette smoke (CS) identified as the main causative agent1. COPD is characterized by progressive obstruction of the airways associated with chronic inflammation of the peripheral airways and progressive destruction of the lung parenchyma2, which contribute to the two major phenotypes of chronic bronchitis and emphysema, each with a different pathophysiology and symptoms3. Chronic inflammation and increased oxidative stress may cause tissue destruction and impair lung maintenance and repair, which are associated with the pathogenesis of emphysema.
It is well known that mechanisms of programmed cell death (i.e., extrinsic and intrinsic apoptosis) contribute to the loss of lung structural cells in the pathogenesis of emphysema4. We and other researchers previously demonstrated the activation of apoptosis signaling pathways in endothelial, epithelial, and fibroblast cells subjected to cigarette smoke extract (CSE) exposure5,6,7. However, the molecular mechanisms of apoptosis and their significance to the etiology of emphysema remain only partially understood.
Recent studies using in vitro and in vivo models of CSE and CS exposure, respectively, as well as human lung tissue from COPD patients, have demonstrated a role for the cellular autophagy pathway (also called macroautophagy) in the pathogenesis of emphysema8,9. Autophagy is a homeostatic process for the turnover of cytoplasmic proteins and organelles. Lung tissue derived from COPD patients or from mice chronically exposed to CS display increased expression levels of autophagy- related proteins and increased autophagosome numbers. A genetic deletion study of crucial autophagy proteins, such as Beclin 1 and microtubule-associated protein-1 light chain-3B (LC3B), revealed the inhibition of CSE-induced lung epithelial cell death in response to CS exposure8.
In addition to the general autophagy pathway, selective forms of autophagy may also be important in the pathogenic mechanisms of COPD. Of these, mitophagy provides a mechanism for the selective autophagic degradation of mitochondria8. According to previous reports, CSE caused mitochondrial dysfunction by decreasing the mitochondrial membrane potential (Δψm) and increasing the production of mitochondrial reactive oxygen species (mtROS). Furthermore, a genetic deficiency experiment of the mitophagy regulator protein, PTEN-induced putative kinase-1 (PINK1), and treatment with the mitophagy/fission inhibitor, Mdivi-1, demonstrated protection against CSE-induced necroptosis and mitochondrial dysfunction in epithelial cells. In lung tissue of COPD patients, lung epithelial cells displayed increased expression levels of PINK1 and the necroptosis protein, receptor-interacting serine/threonine protein kinase 3. These studies have also indicated mitophagy-dependent necroptosis in lung emphysematous changes in response to CS exposure9 and suggest that the activation of mitophagy by CS exposure may promote the induction of necroptosis and lead to depletion of the functional mitochondrial pool during chronic CS exposure9,10,11. Therefore, strategies targeting this pathway may lead to novel therapies for COPD and emphysema. However, the precise mechanism by which mitophagy contributes to lung injury and cell death in the CS exposure model remains unclear. Further studies are needed to improve our understanding of the role of mitophagy in the pathogenesis of emphysema.
Phosphodiesterase-4 (PDE4) inhibitors provide therapeutic benefits, particularly in patients with late-stage inflammation-dominant COPD. Phosphodiesterases (PDEs) are cyclic adenosine monophosphate (cAMP)- or cyclic guanosine monophosphate-specific enzymes that are ubiquitously distributed in most human cells. Eleven PDE isozymes have been identified to date12. Of these, PDE4, which hydrolyzes cAMP, is expressed in all lung structural cells, such as smooth muscle cells, airway epithelium, and inflammatory cells (i.e., neutrophils, lymphocytes, and macrophages)13. Phase III clinical studies have shown that the PDE4 inhibitor roflumilast significantly improved clinical outcomes, such as post-bronchodilator forced expiratory volume in 1 second, and reduced the exacerbation rate and dyspnea severity in the clinical phenotype of chronic bronchitis14. In vivo studies have confirmed that the PDE4 inhibitor roflumilast mitigates emphysema in chronic CS-exposed mice15. In the current study, we examined the functional significance of mitophagy and its relationship with cell death in the context of CSE-induced lung epithelial cell injury. We also investigated the potential therapeutic effects of roflumilast on mitophagy-dependent cell death in CSE-induced Beas-2B cells.
Materials and methods
1. Chemicals and reagents
The MitoSOX Red Mitochondrial Superoxide Indicator Kit was purchased from Life Technologies (Carlsbad, CA, USA). The TMRE Mitochondrial Membrane Potential Assay Kit was obtained from Abcam (Cambridge, UK). Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), and penicillin-streptomycin solution were obtained from Welgene (Gyeongsan, Korea). Dimethyl sulfoxide (DMSO) and 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The following primary antibodies used in this study were purchased from Cell Signaling Technology (Beverly, MA, USA): anti-dynamin-1-like protein (DRP1), anti-phospho-DRP1, anti-PINK1, and anti-actin. The secondary antibodies, anti-rabbithorseradish peroxidase (HRP), and anti-mouse-HRP, were purchased from Santa Cruz Biotechnology (Dallas, TX, USA).
2. Cell culture
Beas-2B human lung epithelial cells were purchased from Lonza Biologics (Cambridge, UK). Cells were cultured at 37℃ in 5% CO2 in DMEM (Welgene) supplemented with 100 U/mL penicillin, 100 µg/mL streptomycin, and 10% FBS (Welgene). Cells were also treated with 0.01% DMSO as a vehicle control. In the indicated experiments, cells were pre-treated with roflumilast (Santa Cruz Biotechnology) for 2 hours and exposed to 10% CSE for the indicated time intervals.
3. Preparation of CSE
Kentucky 3R4F research-reference filtered cigarettes (The Tobacco Research Institute, University of Kentucky, Lexington, KY, USA) were consumed using a peristaltic pump (VWR International, Radnor, PA, USA). The filters were removed from the cigarettes before the experiments. Each cigarette was consumed to a 17-mm butt remaining for 7-8 minutes. The smoke from eight cigarettes was bubbled through 40 mL of cell culture medium, and this solution, regarded as 100% strength CSE, was adjusted to a pH of 7.45 and used within 15 minutes of preparation.
4. Cytotoxicity and viability assays
Lactate dehydrogenase (LDH) release was measured using a cytotoxicity detection kit (Roche, Indianapolis, IN, USA) according to the manufacturer's protocol. After gentle agitation, 100 µL of culture medium was collected at various times and after different doses during the assay. Cell viability was measured using blue formazan, which is metabolized from MTT by mitochondrial dehydrogenases that are active only in living cells. Beas-2B cells were seeded onto 48-well plates (1×104 cells/well) and cultured in DMEM for the indicated time periods. Cells were exposed to 10% CSE for the indicated time intervals. MTT reagent (5 mg/mL) was added to each well, and the plate was incubated for an additional 2 hours at 37℃. The medium was then removed and the intracellular formazan product was dissolved in 300 µL DMSO. The absorbance of each well was measured at 570 nm on a microplate reader (Molecular Devices Emax, Sunnyvale, CA, USA). Optical density values from untreated control cells were designated as 100%.
5. Flow cytometric analysis
mtROS generation was assayed using MitoSOX Red (510/580 nm ex/em ), a mitochondrial superoxide indicator. For the assay, a stock solution of MitoSOX Red was diluted in phosphate-buffered saline (PBS) and added to 10×106 cells/well at a final concentration of 5 µM, and incubated for 10 minutes at 37℃ as described by the manufacturer. Samples were then centrifuged for 5 minutes at 600 ×g, and the pellets were resuspended in 1 mL PBS and subsequently transferred to 5-mL fluorescence-activated cell sorting (FACS) tubes. The TMRE Mitochondrial Membrane Potential Kit uses tetramethylrhodamine, ethyl ester (TMRE; 488/575 nm ex/em) to label active mitochondria. TMRE was added to cells in media at a final concentration of 200 nM and incubated for 20 minutes at 37℃. Samples were then centrifuged for 5 minutes at 600 ×g, and the pellets were suspended in 0.2% bovine serum albumin in PBS and subsequently transferred to 5-mL FACS tubes. For the determination of mtROS and mitochondrial membrane potential (Δψm), the assays were performed on a FACScalibur system (BD Biosciences, San Jose, CA, USA).
6. Confocal microscopy
mtROS levels and mitochondrial membrane potential in CSE-induced Beas-2B cells were measured using MitoSOX Red and TMRE assays as described by the manufacturer. Briefly, cells were incubated in growth medium containing MitoSOX Red at a final concentration of 5 µM for 10 minutes at 37℃, protected from light. Cells were gently washed one time with warm PBS, and the slides were mounted using 4',6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA, USA). For each designated treatment, cells were incubated in culture medium with TMRE at a final concentration of 100 nM for 20 minutes at 37℃, protected from light. Cells were gently washed one time with warm PBS, and the slides were mounted using DAPI. The fluorescence of MitoSOX Red and TMRE was detected under a confocal microscope. Digital images were acquired on an inverted confocal laser-scanning microscope (LSM Pascal, Zeiss, Germany) at 2,048×2,048 pixels. Images were captured using either a 40× or 100× oil immersion objective lens, and the optical section was <1 µm.
7. Western blot analysis
Proteins were extracted from Beas-2B cells using radioim-munoprecipitation assay buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], and 1% NP-40) according to the manufacturer's protocol. Then, 20-40 µg of protein from each sample was separated by 10%-15% SDS--polyacrylamide gel electrophoresis and transferred to a polyvinylidene fluoride membrane (Millipore, Bedford, MA, USA). The membrane was blocked with 5% non-fat skim milk (w/v) for 60 minutes at room temperature and incubated with the indicated primary antibody (anti-cleaved-caspase-3, anti-cleaved-caspas-8, anti-DRP1, anti-phospho-DRP1, anti-PINK1, or anti-β-actin) at 4℃ overnight. The membrane was incubated with a 1:3,000-1:5,000 dilution of HRP-conjugated secondary antibody for 1 hour at room temperature. Each protein was detected using the Western Blot Hyper HRP Substrate Kit (TaKaRa Bio, Shiga, Japan). Band intensity was quantified by densitometric analysis using NIH ImageJ software.
8. Small interfering RNA transfection
Anti-human small interfering RNAs (siRNAs) purchased from Bioneer (Daejeon, Korea) were used for DRP1 and PINK1 knockdown as described by the manufacturer. The target sequences for si-DRP1 included 1040816, 1040813, and 1040810, and those for si-PINK1 included 1116919, 1116915, and 1116923. The latter had the highest inhibition efficiency. Synthetic sequence-scrambled siRNA was used as a negative control (non-targeting) siRNA. Cells were plated and cultured in growth media until cell confluency reached 70%-80% prior to siRNA transfection using Lipofectamine 2000 (Invitrogen, Shanghai, China) according to the manufacturer's instructions. Cells were harvested after 48 hours for Western blot analysis.
Results
1. CSE causes mitochondrial dysfunction and mitophagy in Beas-2B epithelial cells
To investigate the effect of CSE on mitochondrial function in human bronchial epithelial cells (Beas-2B), we evaluated the mitochondrial membrane potential (Δψm) and mtROS production using TMRE and MitoSOX assays, respectively. CSE caused mitochondrial depolarization and a decrease in functional mitochondria in Beas-2B cells (Figure 1A). We also observed that CSE treatment increased mtROS production in Beas-2B cells in a dose-dependent manner (Figure 1A). To investigate whether CSE induces mitophagy in lung epithelial cells, we measured the expression of p-DRP1 and PINK1, regulator proteins of mitophagy. Compared to untreated cells, the expression of p-DRP1 and PINK1 was increased by 16.4- and 5.7-fold, respectively, in Beas-2B cells after CSE treatment (Figure 1B). These results suggest that CSE induces mitophagy in lung bronchial epithelial cells, which is associated with increased expression of mitophagy-related proteins. After CSE exposure, p-DRP1 and PINK1 expression was preferentially increased in the mitochondrial fraction of Beas-2B cells (Figure 1C).
2. CSE treatment in Beas-2B epithelial cells causes mitophagy-regulated apoptotic cell death via upregulation of the mitochondria fission regulator, DRP1, and the mitophagy protein, PINK1
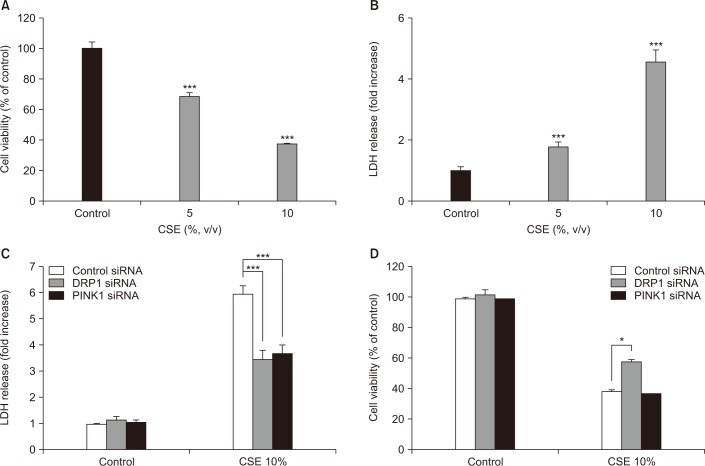
As shown in Figure 2A and B, cell viability, as determined by the MTT assay, and cell death, as determined by the LDH release assay, were effected in a dose-dependent manner after 18 hours of CSE exposure. To determine whether DRP1 and/or PINK1 play a role in CSE-induced cell death, DRP1 or PINK1 was knocked down in Beas-2B cells using DRP1-or PINK1-targeted siRNA. As shown in Figure 2C, control siRNA-transfected cells displayed increased LDH release after CSE treatment (5.71-fold relative to untreated cells), whereas knockdown of DRP1 or PINK1 in Beas-2B cells resulted in decreased LDH release after CSE treatment (3.42- and 3.55-fold, respectively) relative to untreated cells. Moreover, the MTT assay revealed that knockdown of DRP1 decreased cellular cytotoxicity following CSE exposure compared to control siRNA-infected cells, whereas knockdown of PINK1 did not affect cell viability (Figure 2D).
3. Roflumilast inhibited mitochondrial dysfunction and mitophagy in CSE-induced Beas-2B epithelial cells
Next, we investigated the role of roflumilast in mitochondrial dysfunction and mitophagy induced by CSE exposure using confocal microscopy and Western blot analysis. Roflumilast pretreatment markedly protected against mitochondrial dysfunction, as evident by an increase in functional mitochondria and a decrease in mtROS production in CSE-treated cells compared with that observed in epithelial cells treated with CSE only (Figure 3A). Roflumilast also decreased the expression of mitophagy regulator proteins, such as p-DRP1 and PINK1 (Figure 3B).
4. Roflumilast inhibited apoptotic cell death in CSE-induced Beas-2B epithelial cells
Roflumilast markedly decreased CSE-induced cytotoxicity, as measured by the LDH release assay, and increased cell viability, as assessed by the MTT assay (Figure 4A, B). To explore the role of roflumilast in CSE-induced apoptotic cell death, we examined the effect of roflumilast on known apoptosis-related proteins, such as cleaved caspase-3 and -8. Treatment with roflumilast markedly inhibited the expression of these proteins (Figure 3C). The efficacy of roflumilast against CSE-induced cell cytotoxicity was apparent at the concentration of 5 µM roflumilast. DRP1 siRNA-transfected cells showed no more protection following roflumilast treatment in CSE-induced cell death; however, PINK1 siRNA-transfected cells exhibited increased protection following the addition of roflumilast (Figure 4C).
Discussion
Traditionally, chronic inflammation, increased oxidative stress, and/or impaired lung repair processes have been known to contribute to the pathogenesis of emphysema, which is mainly caused by CS and biomass equivalent, and is characterized by an increase in alveolar wall cell death and/or a failure of alveolar wall maintenance. There are no current therapies available that either prevent or cure the progression of emphysema16,17. Preclinical studies have shown that PDE4 inhibitors consistently reduce the accumulation of neutrophils in bronchoalveolar lavage fluid following short-term exposure to CS in mice 18. Thus, PDE4 inhibitors have been exploited primarily for their anti-inflammatory effects in the treatment of COPD, especially in the chronic bronchitis-dominant phenotype. However, several studies that investigated the effects of PDE4 inhibitors on the development of emphysema19 have mainly focused on apoptosis20. We also previously investigated the effects of a PDE4 inhibitor in CSE-induced apoptosis of lung structural cells, and revealed that the PDE4 inhibitor rolipram protects lung fibroblasts from CSE-induced apoptosis by inhibiting the activation of caspase-3 and -821. Autophagy, a cellular pathway for the degradation of damaged organelles and proteins, has gained increasing importance in human diseases, both as a modulator of pathogenesis and as a potential therapeutic target22,23. In addition, autophagy can influence most fundamental processes, including inflammation and immune responses, the host defense, metabolic pathways, and programmed cell death. Some studies have suggested that autophagy promotes lung epithelial cell death, airway dysfunction, and ultimately emphysema in response to CS exposure9,24,25,26. Furthermore, recent studies have reported that CS exposure in epithelial cells promotes the autophagy-dependent turnover of mitochondria (mitophagy) via mitochondrial dysfunction, as evident by a decrease in mitochondrial membrane potential and an increase in mtROS production9. Here, we confirmed that CSE causes mitochondrial dysfunction using TMRE and MitoSOX assays. We also demonstrated that roflumilast markedly inhibited mitochondrial dysfunction by increasing functional mitochondria and decreasing mtROS production.
In general, mitophagy is a cellular response that is commonly associated with mitochondrial dysfunction8. PINK1 and Parkin represent well-recognized regulators of mitophagy in neural cells and tissues27. There are two altered mitochondrial dynamics in terms of mitophagy (i.e., fusion or fission), which play key roles in the response of cells to exogenous stress. Mitochondrial fission, a process regulated by DRP1, is needed to trigger mitophagy28. The phosphorylation of DRP1 promotes DRP1 recruitment to mitochondria29. In adult cardiac myocytes, overexpression of the dominant-negative form of DRP1 results in decreased mitochondrial fission and mitophagy29. Mild oxidative stress specifically triggers mitophagy in a DRP1-dependent manner11. Recent studies have applied Mdivi-1, a pharmacological inhibitor of DRP1, for the functional study of mitophagy30,31,32,33. We confirmed that CSE increased the expression of mitophagy-associated proteins in Beas-2B cells. Furthermore, we showed that the expression levels of p-DRP1 and PINK1 induced by CSE exposure were significantly reduced after pre-treatment with roflumilast. And the MTT assay demonstrated that knockdown of DRP1 decreased cellular cytotoxicity following CSE exposure compared to control siRNA-infected cells, whereas knockdown of PINK1 did not affect cell viability. These inconsistent results of PINK1 need to be elucidated although generally PINK1 promotes cell death in response to CSE.
Many studies have implicated CS-induced apoptosis in alveolar epithelial cells as a possible mechanism for the development of emphysema21,34. CS can induce apoptosis and the production of apoptogenic factors in various structural lung cell types, including alveolar macrophages, bronchial epithelial cells, endothelial cells, and fibroblasts35. In vivo, chronic CS exposure of mice resulted in a significant increase in apoptotic cells in the bronchial and alveolar epithelia36. We previously reported that CSE induced Fas receptor-mediated death-inducing signaling complex formation, the most proximal event in the extrinsic apoptotic pathway6. CSE has been shown to induce both apoptosis and necrotic cell death in airway epithelial cells37. We also found that roflumilast treatment protected against the decrease in viable cells following CSE exposure (Figure 4A). In addition, the expression levels of cleaved caspase-3 and -8 proteins were increased in CSE-induced cells, while treatment with roflumilast markedly decreased the expression of these proteins (Figure 4C). Taken together, these results suggest that roflumilast confers protection against CSE-induced apoptotic cell death in Beas-2B cells. Previous studies have reported a functional role for autophagic proteins in CS-induced epithelial cell death24,25. Cultured epithelial cells exposed to CSE responded with increased autophagosome formation and the accumulation of LC3B-II. CSE induced the extrinsic apoptosis pathway in epithelial cells and the downstream activation of pro-apoptotic caspases. A genetic deletion study of crucial autophagy proteins (i.e., Beclin 1 and LC3B) revealed an inhibition in apoptosis in response to CSE exposure in vitro, suggesting that increased autophagy, to a certain extent, occurred in association with epithelial cell death and also may have cross talk between them24,25. Therefore, an emerging hypothesis is that mitophagy may also contribute to cell death in a context-specific fashion. Although published studies have indicated a relationship between necroptosis and the disintegration of mitochondria (mitophagy), the precise mechanism remains to be elucidated38. Several reports have implicated altered mitochondria as downstream effectors of necrosome activation39,40. In this study, we only investigated the role of roflumilast on mitophagy-dependent cell death, although DRP1-dependent mitophagy in response to CSE can also act as an upstream regulator of apoptotic cell death. DRP1- or PINK1-knockdown cells displayed a significant reduction in cell death in response to CSE exposure relative to control siRNA-transfected cells.
In conclusion, in bronchial epithelial cells, roflumilast diminished CSE-induced cell death by regulating mitophagy-related proteins. Similar to Mdivi-1 (a known inhibitor of mitochondrial fission), roflumilast may confer cytoprotection against CSE by inhibiting mitochondrial fission. These results suggest the potential utility of roflumilast as a preventive therapeutic targeting cell death in addition to its anti-inflammatory action in the treatment of emphysema and COPD.



 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Print
Print Download Citation
Download Citation