Diagnostic Approaches for Idiopathic Pulmonary Fibrosis
Article information

Abstract
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, fibrosing interstitial pneumonia with a very poor prognosis. Accurate diagnosis of IPF is essential for good outcomes but remains a major medical challenge due to variability in clinical presentation and the shortcomings of existing diagnostic tests. Medical history collection is the first and most important step in the IPF diagnosis process; the clinical probability of IPF is high if the suspected patient is 60 years or older, male, and has a history of cigarette smoking. Systemic assessment for connective tissue disease is essential in the initial evaluation of patients with suspected IPF to identify potential causes of interstitial lung disease (ILD). Radiologic examination using high-resolution computed tomography plays a pivotal role in the evaluation of patients with ILD, and prone and expiratory computed tomography images can be considered. If additional tests such as surgical lung biopsy or transbronchial lung cryobiopsy are needed, transbronchial lung cryobiopsy should be considered as an alternative to surgical lung biopsy in medical centers with experience performing this procedure. Diagnosis through multidisciplinary discussion (MDD) is strongly recommended as MDD has become the cornerstone for diagnosis of IPF, and the scope of MDD has expanded to monitoring of disease progression and suggestion of appropriate treatment options.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, fibrosing interstitial pneumonia characterized by progression and poor prognosis, with a median survival of 3 to 5 years [1-3]. Therefore, accurate diagnosis is essential to optimize early treatment for better outcomes in IPF [4,5]. However, accurate diagnosis of IPF remains challenging due to variability in the interpretation of radiopathologic findings and the shortcomings of existing diagnostic tests 6 . Since the first IPF international guidelines were published in 2001, diagnosis of IPF has evolved through a better understanding of the disease mechanism and advances in diagnostic methods including high-resolution computed tomography (HRCT), biopsy techniques, and multidisciplinary discussion (MDD) [7]. Currently, diagnosis of IPF is based on the dynamic integration of clinical, radiologic, and histopathologic information through MDD (Figure 1). The aim of this review is to address the current diagnostic process for IPF based on emerging evidence and the most recent international guidelines.
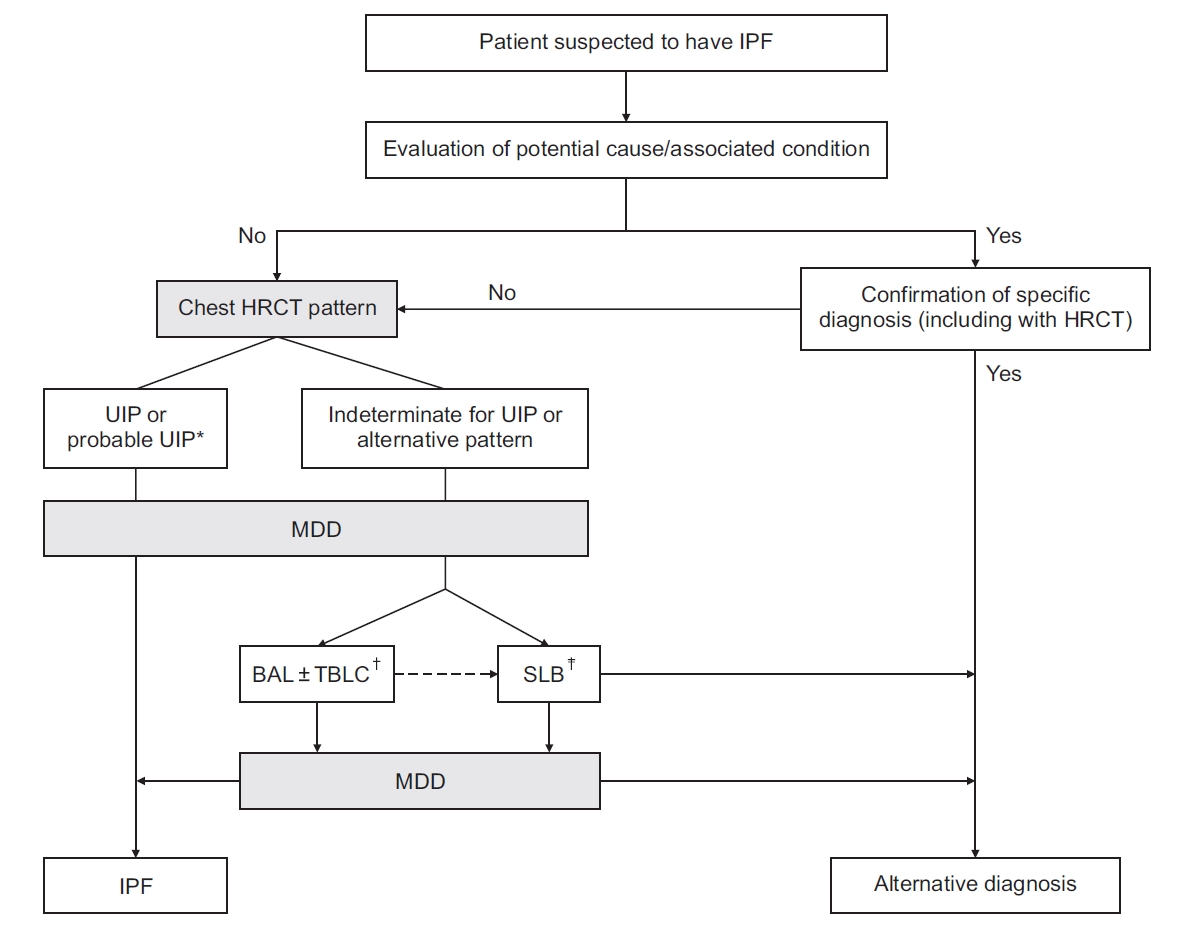
Diagnostic algorithm for idiopathic pulmonary fibrosis (IPF). Adopted from Raghu et al. [17] *Patients with a radiological pattern of probable usual interstitial pneumonia can be diagnosed with IPF after multidisciplinary discussion without confirmation by lung biopsy in the appropriate clinical setting (i.e., 60 years or older, male, smoker). †Transbronchial lung cryobiopsy may be preferred to surgical lung biopsy in centers with appropriate expertise and/ or in some patient populations. ‡Surgical lung biopsy may be justified in some patients with nondiagnostic finding on transbronchial lung cryobiopsy. HRCT: high-resolution computed tomography; UIP: usual interstitial pneumonia; MDD: multidisciplinary discussion; BAL: bronchoalveolar lavage; TBLC: transbronchial lung cryobiopsy; SLB: surgical lung biopsy.
Initial Assessment
Collection of medical history is the first and most important step in the IPF diagnosis process. If interstitial lung disease (ILD) is clinically suspected and identified on HRCT, focused history collection about medication use and environmental exposure at home, work, and other frequently visited places in addition to physical examination should be performed to exclude potential causes of ILD [8]. The clinical probability of IPF is high if the patient is 60 years or older, male, and has a history of cigarette smoking 9 . Common clinical features of patients with IPF are shortness of breath, dry cough, and fatigue. However, these clinical symptoms are nonspecific and are often attributed to other conditions such as airway disease, cancer, and heart disease.
In terms of physical examination, bibasilar inspiratory crackles are commonly detected on chest auscultation, and finger clubbing is present in approximately 50% of patients with IPF [10,11]. Lung function parameters including forced vital capacity, total lung capacity (TLC), and diffusion capacity for carbon monoxide are usually reduced but may be normal in early or subclinical IPF stages. The 6-minute walk test (6MWT) is a reliable tool for practical assessment of functional exercise capacity to determine overall cardiopulmonary reserve [12]. Assessment of total distance and arterial oxygen saturation at the beginning and end of the 6MWT should be measured and used as predictors of mortality [12-15].
Radiologic Assessment
Radiologic examination using HRCT plays a pivotal role in the evaluation of patients with ILD and is routinely performed to diagnose IPF. The Fleischner Society and American Thoracic Society (ATS)/ European Respiratory Society (ERS)/ Japanese Respiratory Society (JRS)/ Latin American Thoracic Association (ALAT) guidelines outline the technical requirements for HRCT to obtain optimal-quality computed tomography (CT) images [16,17]. Thin sections (<2 mm), short rotation times, and high spatial resolution reconstruction are needed. Images should be obtained at full inspiration to TLC [18,19]. Volumetric CT acquisition is preferred over non-contiguous imaging to improve the characterization of patch disease and to delineate the extent of disease. Prone CT images may be considered due to the resolution of atelectasis in the prone position, when there is minimal abnormal opacification of dependent opacification on supine CT images [20]. Prone CT images can enhance the detection of honeycombing and reduce observer variation in IPF diagnosis [21]. Expiratory images can be considered to identify air trapping, which would suggest an alternative diagnosis such as fibrotic hypersensitivity pneumonitis [22].
HRCT Patterns
The ATS/ERS/JRS/ALAT guidelines specify four diagnostic HRCT patterns: usual interstitial pneumonia (UIP), probable UIP, indeterminate UIP, and alternative diagnosis (Table 1) [17].
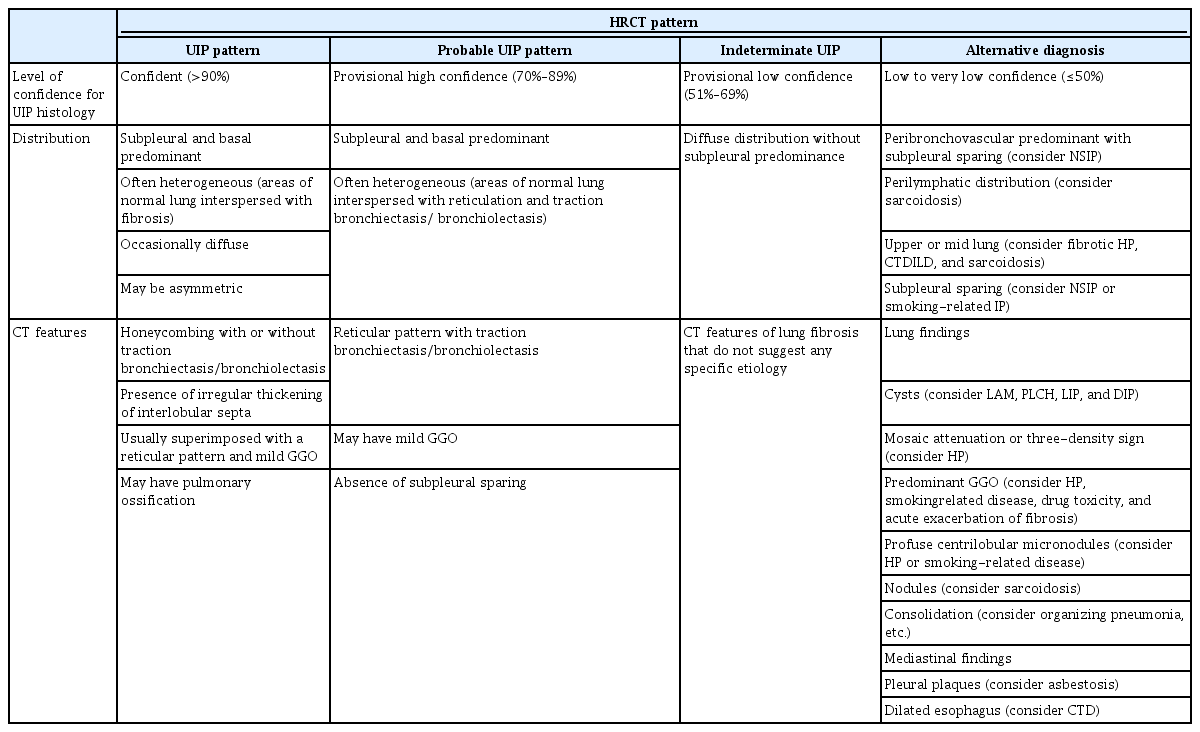
High-resolution computed tomography patterns
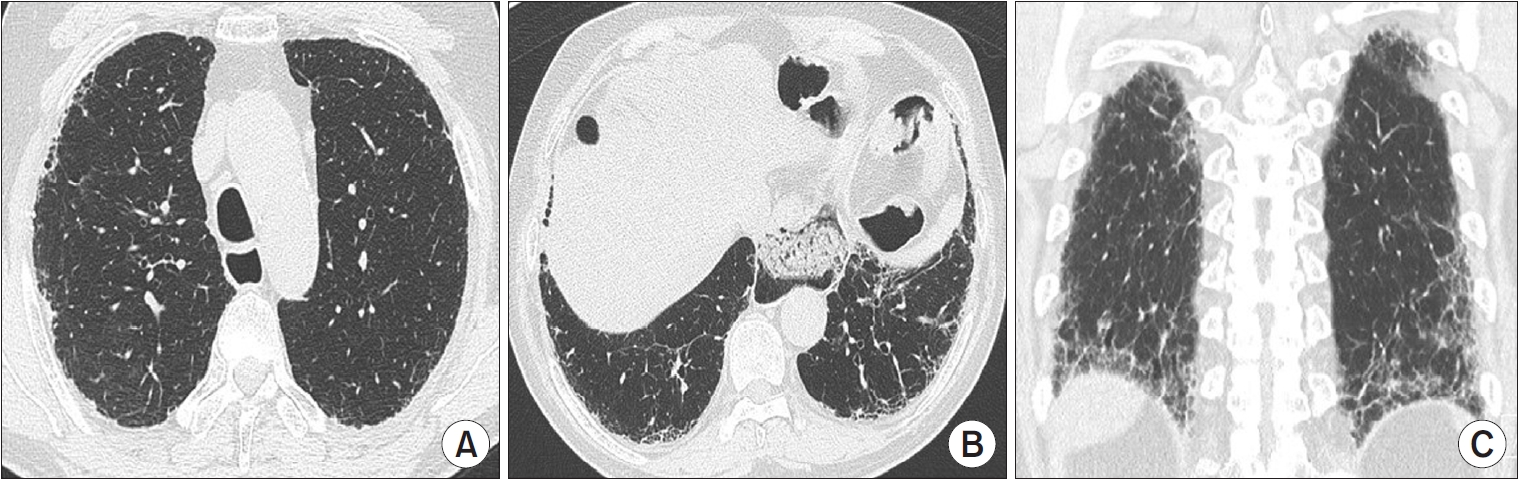
A UIP pattern is the characteristic radiological pattern of IPF. Honeycombing is a necessary and distinguishing feature of a UIP pattern on HRCT (Figure 2). It may be present with or without peripheral traction bronchiectasis or bronchiolectasis. The typical distribution of UIP is subpleural with basal predominance. In a minority of cases, the cranio-caudal distribution of UIP may be relatively uniform, and asymmetric disease may occur in up to 25% of cases [23,24]. In a previous study that investigated the likelihood of histologic confirmation of UIP in patients with a UIP pattern on HRCT, the confidence level of a radiologic UIP pattern for UIP histology was between 90% and 100% [25,26]. Patients with a UIP pattern on HRCT can be diagnosed with IPF without the need for a surgical lung biopsy (SLB) after exclusion of other causes.

Usual interstitial pneumonia pattern on high-resolution computed tomography (HRCT) images. Axial HRCT scans at the upper (A) and the lower (B) lung zones show subpleural reticulation and honeycombing mainly in the lower lobes. Coronal reformation image (C) demonstrates the peripheral distribution of fibrosis and the basal predominance of honeycombing with traction bronchiectasis.
The “possible UIP” category in the 2011 guidelines was updated to “probable UIP” in the 2018 international guidelines because studies showed that selected patients with a “possible UIP” pattern on HRCT are highly likely to have UIP histology, even without evidence of honeycombing on HRCT [27]. Reticular abnormalities with traction bronchiectasis or bronchiolectasis with a subpleural and basal predominant distribution are defined as “probable UIP” (Figure 3). Level of confidence for UIP histology is between 70% and 89% [27-29]. Among patients with a probable UIP pattern on HRCT, those with clinical risk factors (age 60 years or older, male, cigarette smokers) can be diagnosed with IPF after MDD without confirmation by lung biopsy.
Probable usual interstitial pneumonia pattern on high-resolution computed tomography (HRCT) images. Axial HRCT scans at the upper lobes (A) and the lung bases (B) show a bilateral subpleural reticular pattern, which is more severe in the basal lungs. Note the large esophageal hiatal hernia. Coronal reformation image (C) shows subpleural and basal predominant intralobular lines and irregular septal thickening resulting in a reticular pattern, which is associated with traction bronchiolectasis and also mild traction bronchiectasis.
“Indeterminate for UIP pattern” is assigned when HRCT demonstrates features of lung fibrosis that do not suggest any specific diagnosis (Figure 4). In the revised 2022 ATS/ERS/JRS/ALAT guidelines, this category includes CT features of lung fibrosis with a diffuse distribution and without subpleural predominance. The level of confidence for UIP histology is between 51% and 69% [30-32].
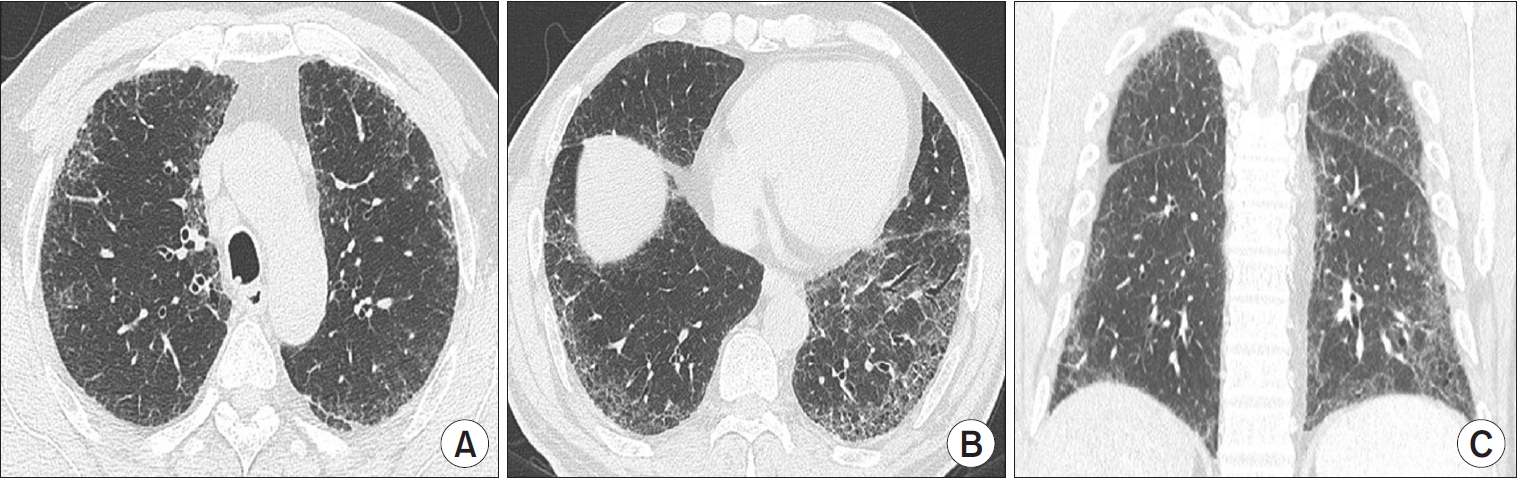
Indeterminate for usual interstitial pneumonia (UIP) pattern on high-resolution computed tomography (HRCT) images. Axial (A, B) and coronal (C) reformation HRCT scans at the upper (A) and lower (B) lung zones show diffuse mild to moderate ground-glass opacities and fine reticulation with no subpleural or craniocaudal predominance. These computed tomography features of lung fibrosis are not suggestive of any specific diagnosis.
Alternative diagnosis pattern on HRCT suggests a diagnosis other than IPF (Figure 5). This may include bronchocentric fibrosis in the upper lobes, profuse mosaic attenuation suggesting hypersensitivity pneumonitis, or extensive ground-glass opacification with subpleural sparing in nonspecific interstitial pneumonia (NSIP). The level of confidence for UIP histology is less than 50% [30,31].
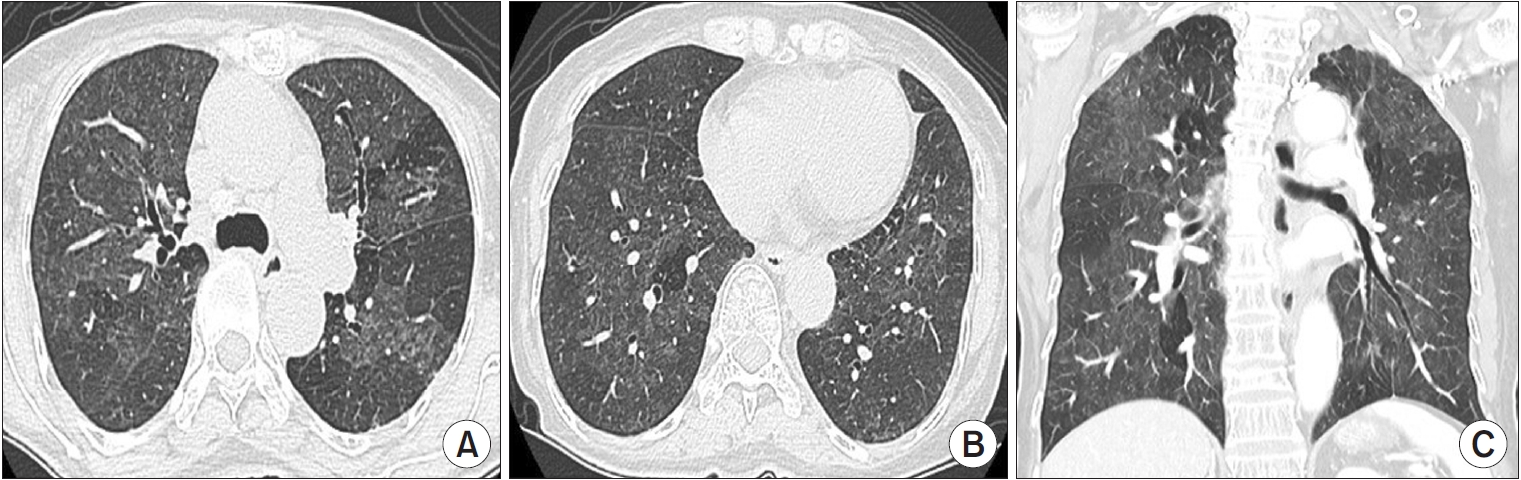
Alternative diagnosis on high-resolution computed tomography (HRCT) images. Axial (A, B) and coronal (C) reformation HRCT scans at the upper (A) and lower (B) lung zones show diffuse ground-glass opacity with poorly defined centrilobular micronodules interposed with areas of normal lung and lobular areas of decreased attenuation. The combination of ground-glass opacity, normal lung, and areas of decreased attenuation gives the lung an appearance suggestive of hypersensitivity pneumonitis.
Rheumatologic Assessment
Systemic assessment for connective tissue disease (CTD) is essential in the initial evaluation of patients with suspected IPF to identify potential causes of ILD. Although there is no clear consensus, autoantibody serologic panels, C-reactive protein, erythrocyte sedimentation rate, antinuclear antibodies, rheumatoid factor, anti-cyclic citrullinated peptide, myositis panel, and muscle enzymes should be evaluated. Current international guidelines recommend that patients be referred for rheumatologic evaluation if they have extrapulmonary disease manifestations suggesting CTD and serologic abnormalities or other characteristics not consistent with IPF (middle-aged, females, non-smokers, or family history of ILD) [17]. In some patients, lung manifestations are the first or only dominant feature of CTD. Some patients also might not fulfil the standard rheumatologic diagnostic criteria at the time of ILD diagnosis, but some of them may be newly diagnosed with CTD during follow-up [33,34]. Therefore, follow-up by a rheumatologist needs to be considered in these patients [34-36].
Bronchoalveolar Lavage
Bronchoalveolar lavage (BAL) is a procedure performed through flexible bronchoscopy to obtain a sample of alveolar cells. BAL findings are usually considered nonspecific for ILD. The percentage of neutrophils in IPF is higher (range, 5.9% to 22.08%) than that in healthy individuals (≤3%) or patients with other ILDs [17,37]. Patients with IPF have a higher proportion of macrophages than those with other ILDs, but the macrophage proportion is lower in individuals with ILDs than healthy individuals (>85%). In the 2011 guidelines, cellular analysis of BAL fluid was not recommended for diagnostic evaluation of IPF due to the additional risk and cost [38]. However, in the 2018 guidelines, BAL fluid cellular analysis is recommended for patients clinically suspected of IPF with an HRCT pattern of probable UIP, indeterminate for UIP, or an alternative diagnosis (conditional recommendation, very low quality of evidence); cellular analysis is not recommended for individuals with a UIP pattern [17,30]. Cellular analysis of BAL fluid may be useful in distinguishing IPF from some alternative ILDs such as eosinophilic pneumonia (eosinophil percentage ≥25%), hypersensitivity pneumonitis (lymphocyte percentage ≥30%), and sarcoidosis (lymphocyte percentage ≥15% and CD4 to CD8 ratio >4:1) [30,39,40].
Histopathologic Examination
UIP is a histopathologic term introduced in 1969 as part of the initial classification of interstitial pneumonia. Histopathologic hallmarks of UIP are the microscopic features of patch dense fibrosis associated with remodeling of lung architecture resulting in honeycomb changes alternating with areas of less affected parenchyma with a mainly subpleural and paraseptal distribution. IPF diagnosis has been based on the histopathologic pattern of UIP; however, there are concerns regarding the risks of SLB and the high frequency of interobserver disagreement in histologic analysis [41]. All patients with suspected IPF do not need surgical biopsy, and the final decision regarding whether to perform this surgery must consider the risks and benefits for individual patients.
Histopathologic Patterns
There are four diagnostic categories of IPF based on HRCT pattern: UIP, probable UIP, indeterminate UIP, and alternative diagnosis (Table 2) [17]. A UIP pattern is defined as patchy, dense fibrosis with the following features: (1) remodeling of lung architecture; (2) honeycomb changes; and (3) distribution that alternates with area of less affected parenchyma, typically in subpleural and paraseptal areas. Probable UIP has some but not all histopathologic features of UIP; features to suggest an alternative diagnosis are absent or only honeycombing may be present. Indeterminate for UIP is defined as fibrosis with or without architectural distortion, favoring either a pattern other than UIP or UIP secondary to another cause. Alternative diagnosis has histologic patterns of idiopathic interstitial pneumonia without fibroblastic foci or loose fibrosis in all biopsies in addition to histologic features indicative of other diseases.
Histopathology patterns and features
Surgical Lung Biopsy
SLB is the gold standard method for histopathological diagnosis of patients with ILD and is indicated when a non-invasive diagnosis is not possible. About one-third of IPF patients in previous studies and clinical trials were diagnosed using SLB [3-5,42]. This method yields adequate specimens for histopathologic diagnosis with a median diagnostic yield around 95% [43]. However, SLB is associated with procedure-related mortality (1.7% and 16% for elective and non-elective SLB), respiratory infections, and delayed wound healing [41]. Definite diagnosis can be obtained in about 90% of patients, but there is a high frequency of interobserver disagreement regarding histopathologic diagnosis [44,45]. Use of SLB to diagnose IPF has decreased over time [46]. However, SLB remains an important diagnostic test to determine adequate treatment through accurate diagnosis in certain patients with ILD.
Transbronchial Lung Cryobiopsy
SLB, including open lung biopsy or video-assisted thoracoscopic lung biopsy, increases patient burden, but transbronchial lung biopsy may not be optimal for diagnosis of IPF because of a small sample size or imaging artifacts [47]. Transbronchial lung cryobiopsy (TBLC) is a new diagnostic approach for diffuse ILDs that is increasingly being used to diagnose IPF. Accumulation of experience over decades and technological advancements have improved diagnostic yield and safety profiles. Recently, the diagnostic yield was reported to be 79%, which is higher than that reported in a previous meta-analysis (72.9%), and can be as high as 85% when three or more samples are collected [48-52]. In terms of complications, reported rates of pneumothorax and pneumothorax requiring chest tube drainage are 9% and 5.6%, respectively, and other severe complications including severe bleeding (1.6%), procedural mortality (0.6%), and exacerbations (1.4%) are uncommon [53-56]. The improved safety profiles compared to previous studies (pneumothorax incidence, 12%; 95% CI, 3% to 21% and moderate/severe bleeding incidence, 39%; 95% CI, 3% to 76%) may be due to accumulation of experience and preventive interventions including endobronchial balloon blockers and fluoroscopic guidance [57]. In the 2020 guidelines of the American College of Chest Physicians and the 2022 ATS/ERS guidelines, TBLC is recommended as an acceptable alternative approach to SLB for histopathologic diagnosis in patients with probable UIP, indeterminate for UIP, or alternative diagnosis on HRCT. Procedural risks, patient preference for TBLC versus SLB, and the experience of the medical center with performing and interpreting TBLC should be considered when deciding to perform TBLC [30,58].
Multidisciplinary Discussion
Given the broad differential diagnosis for IPF, accurate diagnosis is difficult but crucial because of the prognostic and therapeutic implications. Since the joint statement on classification of idiopathic interstitial pneumonia of ATS/ERS in 2002, MDD involving a pulmonologist, a radiologist, and a histopathologist is recommended to diagnose ILD [59]. Evidence in favor of a multidisciplinary approach integrating clinical, radiologic, and histopathologic information has been increasing [60,61]. Walsh et al. [62] investigated 70 patients with ILD and reported that the inter-MDD agreement on diagnosis of IPF (weight–kappa coefficient [kw]=0.71 [interquartile range (IQR), 0.64 to 0.77]) was better than that for idiopathic non-NSIP (kw=0.42 [IQR, 0.37 to 0.49]) and hypersensitivity pneumonitis (kw=0.29 [IQR, 0.24 to 0.40]), and MDD resulted in a diagnosis of IPF with high confidence (77%). Based on these findings, the authors suggested that MDD could also improve interobserver agreement between MDDs for diagnosis of IPF [62]. Therefore, recent guidelines emphasize the importance of dynamic assessment by MDD, and MDD has become the cornerstone for diagnosis and management of ILD [30,36,38]. Despite MDD, however, some patients are not diagnosed and classified with a specific ILD. Therefore, efforts are being made to present confidence levels as a provisional diagnosis in ILD classification during MDD [63]. Originally, MDD was developed and implemented to improve the diagnostic accuracy of ILD, but the scope of MDD has expanded to monitoring of disease progression and determining optimal treatments to achieve better outcomes [64]. Through monitoring of disease progression, MDD can propose changes in treatment including participation in clinical trials, timely implementation of non-pharmacological therapies (supplemental oxygen or pulmonary rehabilitation), and preparation for lung transplantation, resulting in a better prognosis.
Conclusion
Accurate diagnosis of IPF remains a major medical challenge due to variability in clinical presentation and limitations of current diagnostic tests. For better outcomes, a detailed medical history including systemic assessment for connective tissue disease is the first and most important step. Radiologic examination using HRCT plays a pivotal role in the evaluation of patients with ILD. If histopathologic samples are needed, SLB or TBLC may be considered. MDD has become the cornerstone of diagnosis of IPF, and the scope of MDD now includes monitoring of disease progression and suggestion of appropriate treatment options.
Notes
Authors’ Contributions
Conceptualization: Song JW. Methodology: Song JW. Formal analysis: all authors. Data curation: Lee JH. Software: all authors. Validation: Song JW. Writing - original draft preparation: Lee JH. Writing - review and editing: all authors. Approval of final manuscript: all authors.
Conflicts of Interest
Jin Woo Song is an editorial board member of the journal, but he was not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflicts of interest.
Funding
This study was supported by grants from the Basic Science Research Program (NRF-2022R1A2B5B0200 1602) and the Bio & Medical Technology Development Program (NRF-2022M3A9E4082647) of the National Research Foundation of Korea (NRF) funded by the Ministry of Science & ICT, Republic of Korea, as well as by grants from the National Institute of Health research project (2021ER120701) and the Korea Environment Industry & Technology Institute through the Core Technology Development Project for Environmental Diseases Prevention and Management Program funded by the Korea Ministry of the Environment (ARQ202201450001), Republic of Korea.