Immune Evasion of G-CSF and GM-CSF in Lung Cancer
Article information

Abstract
Tumor immune evasion is a complex process that involves various mechanisms, such as antigen recognition restriction, immune system suppression, and T cell exhaustion. The tumor microenvironment contains various immune cells involved in immune evasion. Recent studies have demonstrated that granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF) induce immune evasion in lung cancer by modulating neutrophils and myeloid-derived suppressor cells. Here we describe the origin and function of G-CSF and GM-CSF, particularly their role in immune evasion in lung cancer. In addition, their effects on programmed death-ligand 1 expression and clinical implications are discussed.
Introduction
Immune evasion hinders the effective treatment of lung cancer with immune checkpoint inhibitors. Programmed death-ligand 1 (PD-L1) binds to programmed cell death-1 (PD-1) receptors on cytotoxic T cells to inhibit T cells and decrease cytokine production [1-3]. In addition, neutrophils and myeloid-derived suppressor cells (MDSCs) inhibit immune responses within the tumor microenvironment. Recent studies have demonstrated that granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF) induce immune evasion through various mechanisms [4-10]. Clinical studies have demonstrated that high levels of G-CSF and GM-CSF are associated with a poor prognosis [11-13]. Moreover, exogenous GM-CSF induces PD-L1 expression and promotes tumor progression [8,10,14,15].
Here we describe the origin and functions of G-CSF and GM-CSF in lung cancer, focusing on their role in modulating neutrophils, MDSCs, and PD-L1 expression.
Origin and Function of G-CSF/GM-CSF
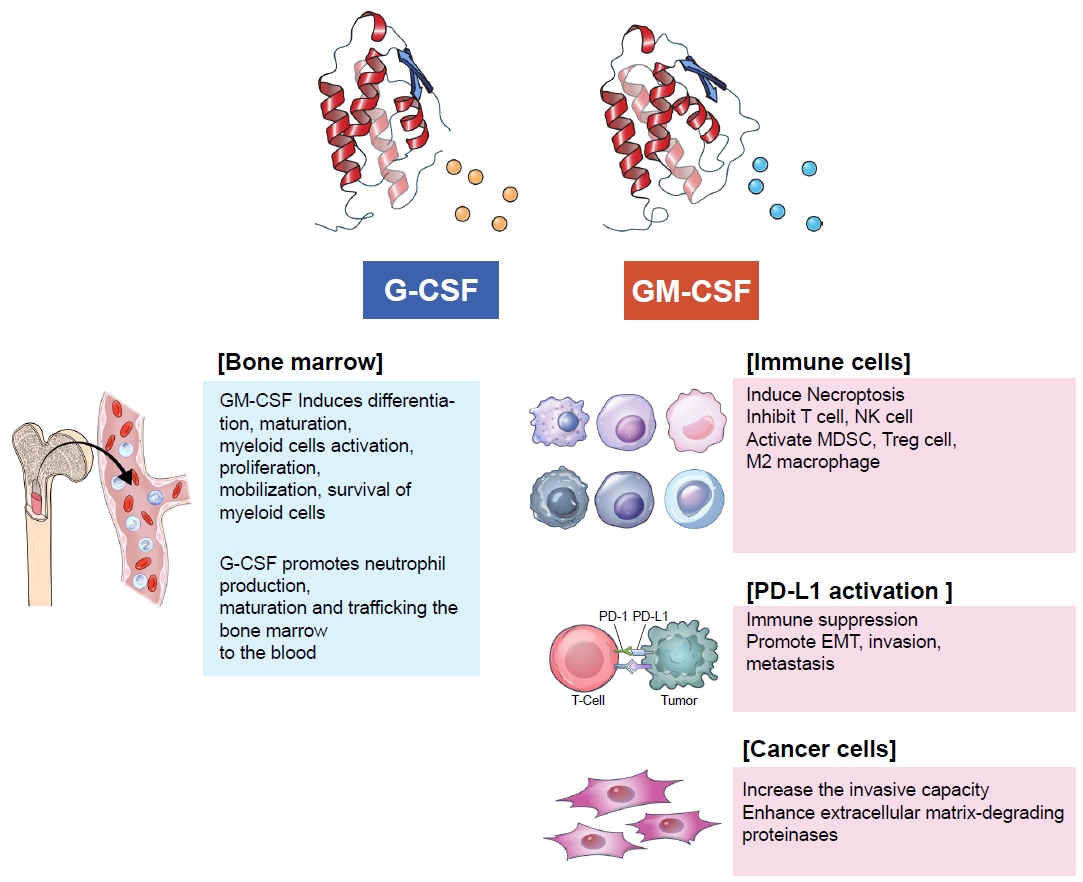
G-CSF and GM-CSF are essential growth factors for hematopoiesis. GM-CSF causes the proliferation and differentiation of myeloid cells, whereas G-CSF regulates neutrophil production, maturation, and mobilization. Vascular endothelial cells, fibroblasts, and immune cells, such as T cells, B cells, macrophages, natural killer cells, and mast cells, produce GM-CSF in response to inflammatory cytokines and the innate immune response [16]. In turn, GM-CSF affects myeloid cell differentiation, maturation, activation, proliferation, mobilization, and survival [17]. Conversely, G-CSF promotes the production, maturation, and migration of neutrophils from the bone marrow to the bloodstream [18]. Clinicians frequently use recombinant G-CSF and GM-CSF to increase the absolute neutrophil count in cancer patients who develop neutropenia following chemotherapy.
Immune Evasion in Lung Cancer by G-CSF/GM-CSF by Modulating Neutrophils, Macrophages and MDSCs
Lung cancer cells secrete G-CSF and GM-CSF to evade the immune system in response to oncogenic and immunologic signals [19]. Tumor-derived G-CSF reprograms myeloid differentiation and produces immunosuppressive neutrophils (Figure 1) [20,21]. Neutrophils play a major role in innate immunity against bacterial infections. In the bone marrow, myeloblasts differentiate into promyelocytes, neutrophilic myelocytes, neutrophilic metamyelocytes, neutrophil stab cells, and segmented neutrophils. Mature segmented neutrophils differentiate into small, high-density neutrophils with anti-tumor effects, whereas undifferentiated neutrophils are large, low-density neutrophils with pro-tumorous effects [22]. Immature neutrophils typically remain in the bone marrow under normal conditions. However, in cancer patients, stimulated low-density neutrophils migrate from the bone marrow to the blood. MDSCs are low-density neutrophils [23]; their levels in the blood are increased by cancer-derived endogenous and exogenous G-CSF and GM-CSF [24]. MDSCs consist of heterogeneous precursors of dendritic cells, macrophages, and granulocytes. Recent studies have demonstrated that 70% to 80% of MDSCs are polymorphonuclear cells, and 20% to 30% are monocytic cells [25]. These cells cause immune suppression in the tumor microenvironment, presenting a significant barrier to cancer immunotherapy [26]. They inhibit the CD8-positive cytotoxic T cells, thereby inhibiting the effects of immune checkpoint inhibitors. In addition, prostaglandin E2 derived from MDSCs can accumulate cancer stem cells and induce PD-L1 overexpression, resulting in immune suppression [27].
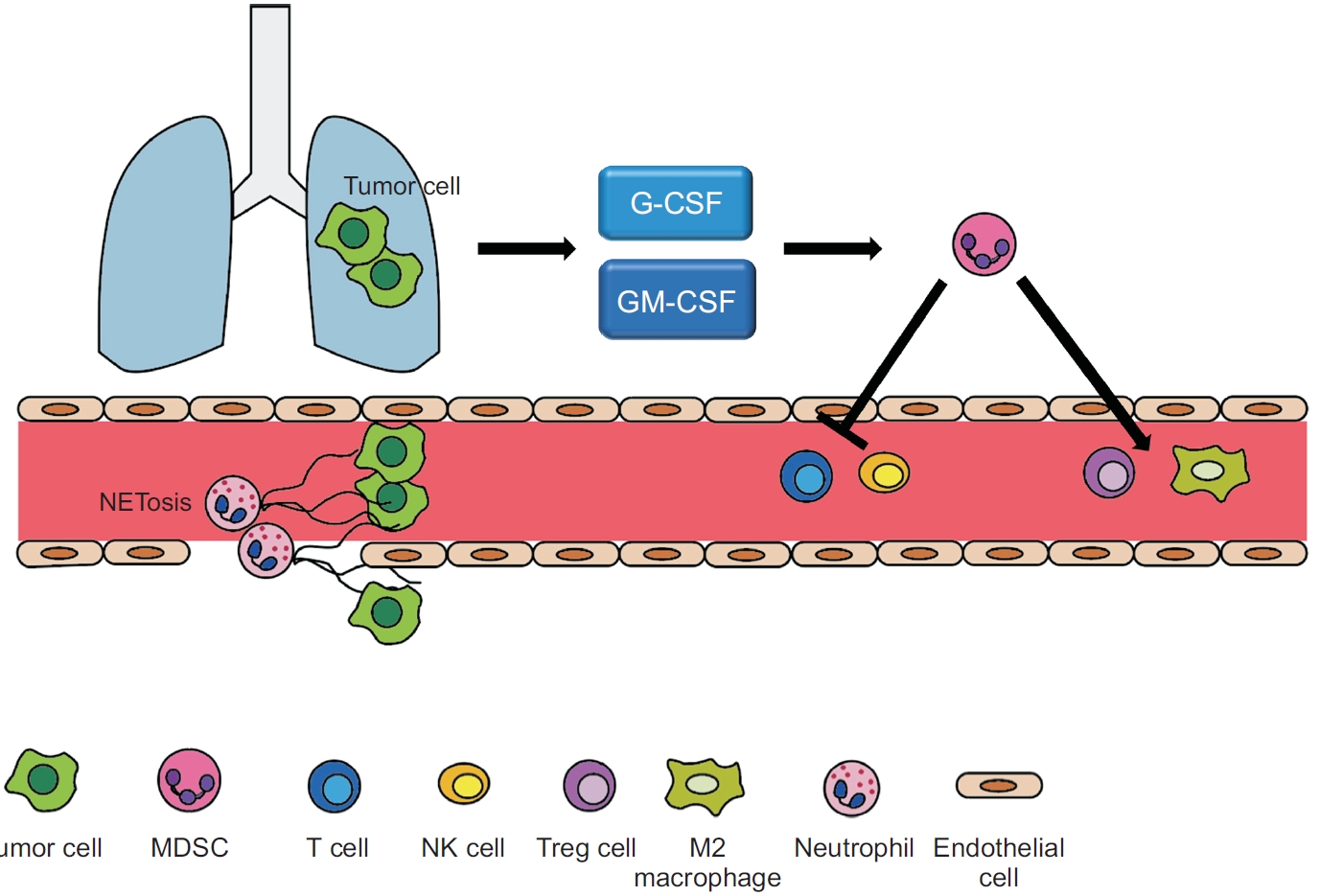
Schematic diagram of immune evasion by granulocyte colony-stimulating factor (G-CSF) and granulocytemacrophage colony-stimulating factor (GM-CSF) in lung cancer. Lung cancer cells secrete G-CSF and GM-CSF, which modulate myeloid differentiation, produce immunosuppressive neutrophils, and induce the accumulation of myeloidderived suppressor cells (MDSCs) in circulation. In turn, MDSCs induce the production of Tregs and differentiation of M2 macrophages, reduce natural killer cell activity, and inhibit T cell proliferation and migration. In addition, neutrophil extracellular trap (NET)-associated granule proteins promote tumor metastasis by secreting neutrophil elastase and matrix metalloproteases. NK: natural killer.
Tumor-associated macrophages (TAMs) are the key cells that create an immunosuppressive tumor microenvironment by producing cytokines, chemokines, growth factors, and triggering the release of inhibitory immune checkpoint proteins in T cells. TAMs can display very different and even opposing phenotypes, depending on the microenvironment in which they are embedded. Macrophages are divided into two main categories: classical activated M1 macrophages (major histocompatibility complex [MHC]-II+CD68+) and alternatively activated M2 macrophages (CD163+CD206+). M1 macrophages have an anti-tumor function, as they are responsible for killing tumor cells through tumor-killing molecules or antibody-dependent cell-mediated cytotoxicity. In contrast, M2 macrophages promote the proliferation and metastasis of tumor cells by expressing various growth factors such as vascular endothelial growth factor, transforming growth factor-β1 (TGF-β1), epithelial growth factor, and hepatocyte growth factor (HGF) [7,28,29].
In human triple-negative breast cancer, high G-CSF expression is significantly associated with CD163+ TAMs and decreased overall survival. In vitro and in vivo data using a panel of human breast cancer and mouse mammary tumor cell lines have demonstrated that tumor-derived GM-CSF is an important cytokine involved in the activation of signal transducer and activator of transcription 5 (STAT5) signaling in macrophages [30].
There are two distinct types of neutrophils, N1 and N2 tumor-associated neutrophils (TANs). N1 TANs inhibit tumor growth and metastasis through antibody-dependent or direct cytotoxicity and activation of various innate and adaptive immune cells [31-33]. Additionally, N1 TANs produces reactive oxygen species through enhanced nicotinamide adenine dinucleotide phosphate hydrogen oxidase activity, thereby exerting a cytotoxic effect on tumor cells [34]. Conversely, N2 TANs promote tumor progression through multiple mechanisms. N2 TANs secrete several enzymes, such as myeloperoxidase, neutrophil elastase (NE), neutrophil collagenase (matrix metallopeptidase 8 [MMP8]), and MMP9, which promote extracellular matrix remodeling and angiogenesis, resulting in tumor proliferation and promote migration [35,36]. Moreover, N2 TANs inhibit the function of effector T lymphocytes by recruiting Tregs via CC motif chemokine ligand 17 (CCL17) secretion [37]. TGF-β is known to be an important cytokine involved in the skewing of neutrophil differentiation toward N2 TANs [33,38,39], while treatment with TGF-β blockade and interferon-β showed a shift toward N1 TANs [40,41].
Neutrophils release an intracellular web-like chromatic structure called neutrophil extracellular traps (NETs), which induce an immune response against infections, particularly by large pathogens [42]. These are released through plasma membrane rupture or expel nuclear chromatin without plasma membrane rupture. NET-associated granule proteins promote tumor metastasis by releasing NE and matrix metalloproteases. These proteases cause cancer cells to undergo metastasis [43].
Tumor-derived G-CSF/GM-CSF induces activation and proliferation of TANs. G-CSF secreted from cancer cells induced TANs to form NETs [35]. Hepatocellular carcinoma-derived GM-CSF stimulated cancer cell migration and invasion by enabling TANs to produce HGF and activating the HGF/c-Met axis [44]. Bv8 may function to modulate or amplify neutrophil mobilization stimulated by G-CSF, through paracrine or autocrine mechanisms [45]. Tumor-derived G-CSF activates a myeloid differentiation program, resulting in increased immunosuppressive neutrophils that possess T cell-suppressive, and retinoblastoma protein (Rb1)low phenotype in an oncogene-driven murine breast cancer model [20].
In addition, G-CSF causes tumor growth, metastasis, and chemotherapy resistance [46-49]. Several studies have shown that a high G-CSF level is associated with thrombosis and a poor prognosis [50].
Effects of G-CSF/GM-CSF on PD-L1
Some patients develop remarkable progression of extra-nodal natural killer/T cell lymphoma (ENKTL) after GM-CSF treatment. GM-CSF treatment significantly increases the expression of PD-L1 mRNA and protein in ENKTL cells. Because PD-L1 has immunosuppressive functions, GM-CSF treatment results in the loss of tumor immune surveillance in ENKTL patients, accelerating cancer cell progression. GM-CSF stimulates PD-L1 expression through the Janus kinase 2 (JAK2)-STAT5 signaling pathway in ENKTL [10].
PD-L1 expression is regulated by multiple signaling pathways, such as MYC, Kirsten rat sarcoma virus (KRAS), STAT3, JUN, phosphatase and tensin homolog (PTEN), and epidermal growth factor receptor (EGFR), which are responsible for the constitutive expression of PD-L1 [51]. In addition, interferon-γ, interleukin-6, interleukin-27, tumor necrosis factor-α, and epidermal growth factor induce PD-L1 expression [52]. The interaction between PD-L1 and PD-1 activates downstream signaling pathways, such as phosphoinositide-3-kinase (PI3K), Linker for activation of T cells (LAT), and Src homology region 2-containing protein tyrosine phosphatase 2 (SHP2), in T cells, which inhibits T cell activation and cytokine production [53]. Immune checkpoint inhibitors, including pembrolizumab, nivolumab, and atezolizumab, reactivate cytotoxic T cells and overcome immune evasion. In a recent in vitro study using lung adenocarcinoma cell lines and human monocyte-derived macrophages, it was suggested that cancer cell-derived GM-CSF induces PD-L1 overexpression on TAMs through the STAT3 pathway (Figure 2) [54].
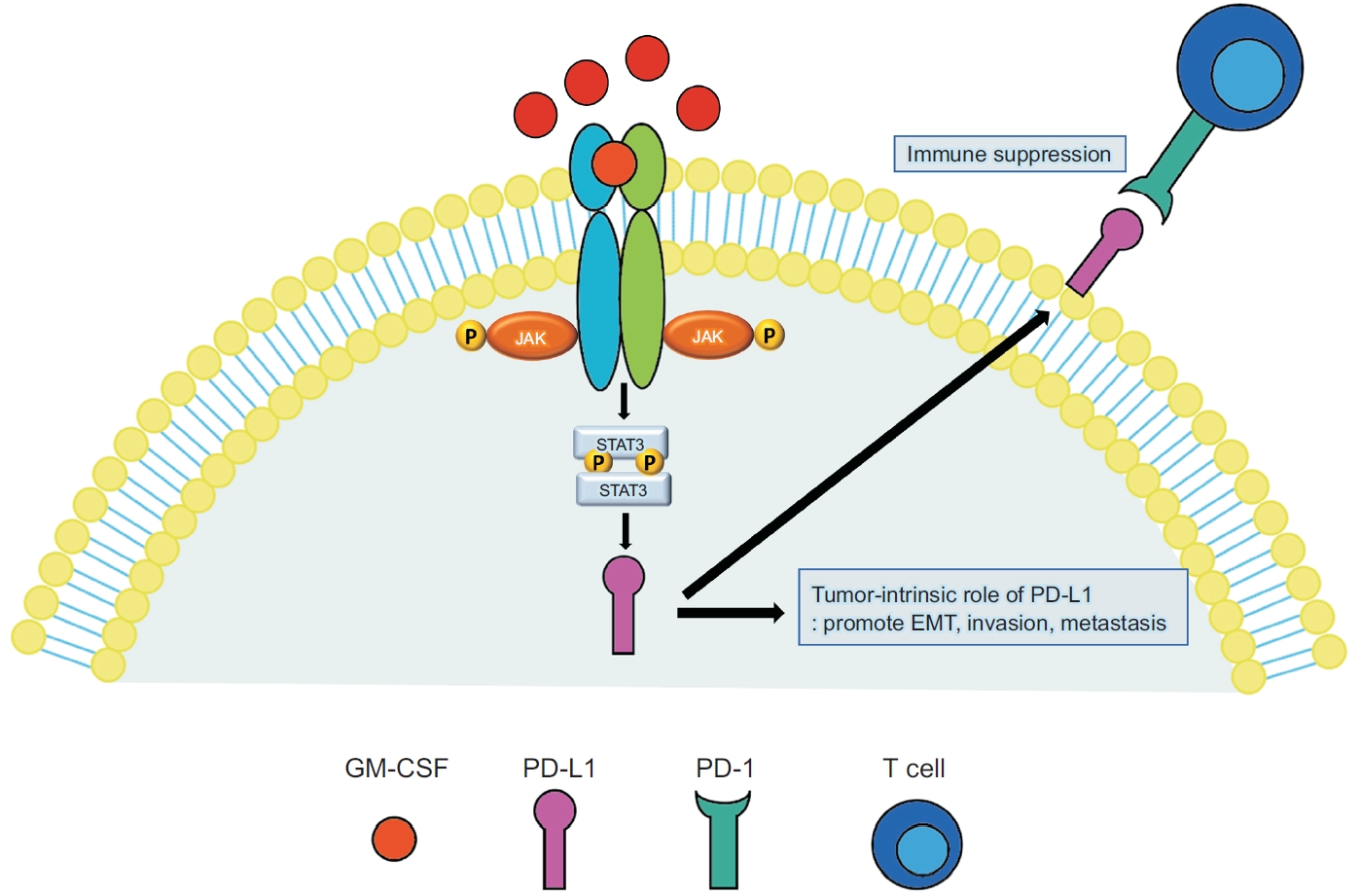
Illustration of the immune evasion mechanism by which granulocyte-macrophage colony-stimulating factor (GM-CSF) increases programmed death-ligand 1 (PD-L1) expression on macrophage through the Janus kinase/signal transducer and activator of transcription (JAK-STAT3) signaling pathway in lung adenocarcinoma. PD-L1 upregulation causes immune suppression by inhibiting T cell activation in a programmed cell death-1 (PD-1)-dependent manner. In addition, PD-L1 promotes cancer cell proliferation, metastasis, and epithelial-mesenchymal transition. This figure highlights the potential therapeutic use of the JAK-STAT3-PD-L1 axis in lung adenocarcinoma. EMT: epithelial-mesenchymal transition.
Recent studies have demonstrated that PD-L1 has intrinsic functions independent of PD-1 [55-57]; it promotes cancer stemness, epithelial-mesenchymal transition, and drug resistance in cancer cells. PD-L1 contains RMLDVEKC and DTSSK motifs in the cytoplasmic domain, which inhibit interferon-mediated cytotoxicity in cancer cells by preventing STAT3 activation and subsequent caspase-7-mediated apoptosis [55]. A recent study demonstrated that PD-L1 was translocated to the nucleus after deacetylation [58]. Nuclear PD-L1 modulates immune response gene expression and consequently decreases the effects of immune checkpoint inhibitors. Therefore, the prevention PD-L1 translocation is a novel strategy for improving the outcomes of immunotherapy in cancer. Because G-CSF enhances PD-L1 expression in cancer, G-CSF blockade may be used as a novel adjuvant treatment with immunotherapy. In the results of a preclinical study using a murine Lewis lung carcinoma cell line, it was shown that blocking GM-CSF can significantly inhibit tumor development. Although anti-GM-CSF therapy did not affect PD-L1 expression in tumor tissues, it suppressed the infiltration and maturation of TAMs and increased T cell infiltration. Although further studies are still required, cancer-derived GM-CSF may be a promising target for anti-cancer therapy [54].
Miyazawa et al. [59] showed that PD-L1 expression is affected by matrix stiffness in lung cancer and that it is much higher in a stiff than a soft matrix. Although the exact mechanism by which matrix stiffness affects PD- L1 expression remains unknown, mechanosensitive Yes-associated proteins may be involved, as they regulate PD-L1 transcription. Some studies have shown that GM-CSF regulates the extracellular matrix by controlling the metabolism of vascular collagens [60]. It also stimulates the proliferation and migration of vascular endothelial cells [61]. Therefore, GM-CSF can cause immune evasion by altering PD-L1 expression and extracellular matrix stiffness.
Clinical Uses of G-CSF and GM-CSF
G-CSF/GM-CSF has been widely used as supportive care for patients receiving chemotherapy. The use of G-CSF/GM-CSF has shown to reduce the incidence of chemotherapy-associated neutropenia by 37% and decrease the duration and severity of neutropenia in patients undergoing cancer chemotherapy [62]. However, recent reports have indicated that overexpression of G-CSF/GM-CSF is associated with poor prognosis in various types of cancers. In a clinical study, G-CSF level was significantly higher in patients who died than in those who survived gastric cancer [63]. In addition, a high G-CSF level was negatively correlated with overall survival. Furthermore, the G-CSF level was significantly higher in a mammary cancer model than in controls, and G-CSF blockade slowed tumor growth. Tumor-derived G-CSF increases tumor growth through MDSC-related immune reactions [24]. Serum levels of GM-CSF in patients with glioblastoma are more than two-fold higher than in healthy controls [64]. Patients with non-small cell lung cancer have higher GM-CSF levels compared to healthy controls. Therefore, GM-CSF is a potential diagnostic marker for non-small cell lung cancer [65]. G-CSF and GM-CSF upregulate the invasive capacity of human lung cancer cells by enhancing the production of extracellular matrix-degrading proteinases [19]. A clinical study of non-small cell lung cancer patients demonstrated that higher GM-CSF levels are positively correlated with poor prognosis [66].
The ongoing clinical trials of G-CSF/GM-CSF in lung cancer are described in Table 1. Apart from one study using pegylated G-CSF, most studies utilized recombinant human GM-CSF, such as sargramostim. Moreover, these studies primarily investigate the use of G-CSF/GM-CSF in combination with other drugs for supportive care purposes, rather than confirming their therapeutic effects.

Ongoing clinical trials of G-CSF/GM-CSF in lung cancer
Interestingly, an in vivo study using a breast cancer model demonstrated that long-term exposure to high levels of G-CSF increased metastasis, while short-term G-CSF administration in conjunction with cytotoxic chemotherapy did not lead to increased metastasis [67]. Therefore, caution should be exercised in the use of recombinant G-CSF/GM-CSF as it has the potential to promote tumor progression and metastasis [17].
Conclusion
Tumor-derived or recombinant G-CSF and GM-CSF have diverse roles in the tumor microenvironment. They disrupt granulopoiesis and neutrophil maturation and increase TANs, particularly pro-tumor N2 neutrophils, which promote tumor growth, invasion, and metastasis. Furthermore, they mobilize immunosuppressive MDSCs into circulation, where MDSCs suppress natural killer and cytotoxic T cells. GM-CSF stimulates PD-L1 expression, which has immunological and PD-1-independent intrinsic functions. Because high G-CSF and GM-CSF levels are associated with poor prognosis in cancer patients, recombinant G-CSF and GM-CSF should be used cautiously.
Notes
Authors’ Contributions
Conceptualization: Chung C. Writing - original draft preparation: all authors. Writing - review and editing: Park YH. Approval of final manuscript: all authors.
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Funding
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIT) (No. NRF-2017R1A5A2015385, NRF- 2022R1A2C2010148) and a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Korea (grant number: HR20C0025).