Introduction
The Korea Interstitial Lung Disease Study Group made a Korean nationwide idiopathic pulmonary fibrosis (IPF) registry in 2008, which analyzed IPF patients diagnosed from 2003 to 2007 [1]. It reported characteristics of Korean IPF patients, showing that old age, lower forced vital capacity (FVC) and diffusing capacity for carbon monoxide (DLco), and higher gender-age-physiology (GAP) index were associated with poor prognosis.
The diagnosis and treatment of IPF had some changes during the recent decade. Although IPF is still a fatal disease characterized by chronic progressive course and limited effective therapies, two antifibrotic agents, pirfenidone and nintedanib, have been shown to be able to slow the decline of lung function [2,3]. Moreover, new guidelines for diagnosis and management of IPF were published [4,5]. Therefore, some changes in diagnosis, treatment, and outcomes during the past decade were expected.
According to randomized clinical trials[2,3] and their extension study[6], the natural history of IPF and treatment effects have been revealed. However, randomized clinical trials have strict inclusion and exclusion criteria. Therefore, results of these clinical trials could not be applied to a lot of patients because they did not meet the inclusion criteria. A registry can bridge the gap between real-world clinical practice and clinical trial. Because real-world data can be more representative of clinical practice than clinical trials although lots of confounding variables were observed and the results should be analyzed with caution. Furthermore, a registry also provides important long-term clinical data on the course of IPF and the impact of new treatments.
This study aimed to describe demographic and clinical characteristics of recent Korean IPF patients and investigate the difference between this registry and the previous Korean registry made in 2008 or registries of other countries.
Materials and Methods
This is a nationwide, investigator-initiated observational study. A total of 23 universities and teaching hospitals were involved in making the new IPF registry from June 2016. Both newly diagnosed IPF and following patients diagnosed with IPF after the previous registry in 2008 were enrolled in this study. In the case of previously diagnosed IPF patients, their baseline characteristics were retrospectively reviewed at the time of initial diagnosis. Patients were diagnosed with IPF according to international guideline [5] at the study site. Medical records of patients were reviewed to fill up the Korean interstitial lung disease web-based registry (http://ipf.crf.kr). For analysis of treatment and survival, only patients diagnosed after 2016 were included because antifibrotic agents started to be covered by medical insurance of Korea in October 2015. Those treated with antifibrotics for more than three months were classified into the antifibrotics group. Patients who were not treated or treated with antifibrotics for less than three months were classified into the non-antifibrotics group. Written informed consent was obtained from all patients before registration except for those who were already dead or lost a follow-up. The registry had no explicit exclusion criteria.
Statistical analysis was performed using SPSS version 24 (IBM Corp., Armonk, NY, USA). Categorical variables are presented as absolute numbers and relative frequencies. Continuous variables are shown as mean±standard deviation. Kaplan-Meier survival analysis and log-rank test were used for survival analysis. Additionally, Cox regression analysis was used for evaluating risk factors for mortality. One-way analysis of variance (ANOVA) was performed to detect differences among patients with different treatments, followed by Tukey’s post-hoc test. This study protocol was reviewed and approved by the Institutional Review Board of Ulsan University Hospital (approval number: 2015-07-046) and registered with ClinicalTrials.gov (NCT04160715).
Results
1. Baseline clinical characteristics
A total of 2,424 IPF patients were registered from June 2016 to August 2018. Of them, 285 patients without basic information such as age, sex, diagnosis date, or final follow-up date were excluded. Thus, 2,139 patients were finally included in the analysis. Their baseline characteristics are outlined in Table 1. Patients were predominantly males (76.1%) with a mean age of 67.4±9.3 years. Smoking history was reported by 71.0% of patients. At the time of diagnosis, 12.9% continued to smoke. A family history of interstitial lung diseases was reported by 3.4% of patients.
Occupational and environmental exposure are shown in Table 2. A total of 1,775 patients (83.0%) have reported their occupational status. Ten percent of patients worked in the industrial field and 6.5% worked in the construction field. Primary industry including agriculture, fishing, and livestock was reported by 7.5% of patients. Dust exposure was reported by 534 patients (25.0%). The most commonly exposed dust was stone dust.
2. Initial symptoms and diagnostic procedures
Table 3 displays initial symptoms and diagnostic tests. At the time of diagnosis, 308 patients (14.4%) were asymptomatic. Among those patients with symptoms, the most common symptom was dyspnea (52.0%), followed by cough (50.4%). The mean modified Medical Research Council dyspnea scale was 1.4±1.0. Among initial diagnostic workups, autoantibody test for connective tissue was performed for 1,092 patients (68.7%). In addition, bronchoscopy with bronchoalveolar lavage was performed for 815 patients (38.1%) and echocardiography was performed for 1,006 patients (47.0%).
A 6-minute work test was performed for 1,088 patients (50.9%). The mean walk distance was 399±182 m. The minimum oxygen saturation was 90.3%±14.1%. The mean GAP index was 3.4±1.5. At the time of diagnosis, 56.9%, 32.7%, 10.4% of patients were at GAP stage 1, 2, and 3, respectively.
A total of 448 patients (20.9%) were diagnosed after surgical lung biopsy. The proportion of surgical lung biopsy was decreased over time: 31.9% before 2012, 20.5% between 2012 and 2015, and 15.3% since 2016 for all IPF patients.
3. Pulmonary function and chest computed tomography findings
Table 3 also shows initial pulmonary function test and chest computed tomography (CT) findings. Pulmonary function tests and high-resolution computed tomography (HRCT) have been performed for almost all patients. Initial FVC was 74.6±17.5% predicted and DLco was 63.6%±22.0% predicted.
On initial chest CT, honeycombing was the most common finding. It was found in 67.0% of patients. Emphysema was combined in 32.7% of patients. The definite radiological usual interstitial pneumonia (UIP) pattern was found in 45.4% of patients. Probable UIP pattern was found in 42.4% of patients. In 5.8% of patients, inconsistent with UIP pattern was found. Lung cancer was diagnosed simultaneously with IPF in 3.4% of patients.
4. Treatment
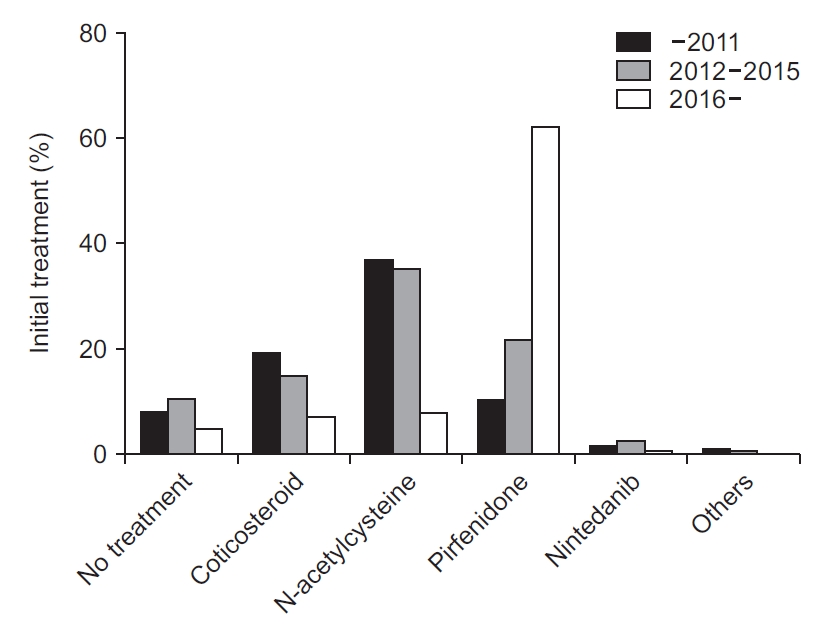
Pirfenidone was the most frequently prescribed drug initially (31.9%), followed by N-acetylcysteine (27.8%) and corticosteroid (13.7%) (Table 4). Nintedanib, another antifibrotic agent, was prescribed in only 2.2% of patients initially. At first, 187 (8.7%) patients were not treated. When the period was divided into three: before 2012, 2012 to 2015, and since 2016, the frequency of choosing pirfenidone as a first treatment increased over time (Figure 1). It was prescribed in 10.6% of the patients before 2012 and 62.4% since 2016 as the first treatment. The frequency of selection of N-acetylcysteine as the first treatment markedly decreased after having pirfenidone under the insurance coverage in late 2015. It was the most frequently prescribed drug before 2016. Over 35% of the patients were treated with N-acetylcysteine initially. However, it was prescribed in only 8.1% of patients since 2016. The use of corticosteroids also gradually diminished.
The proportion of treatment with antifibrotic agents for more than three months also increased. Before 2012, only 20.4% of patients were treated with antifibrotics for more than three months. This proportion increased to 44.4% between 2012 and 2015. It then increased to 79.2% since 2016 (Figure 2).
5. Prognosis
About one-third of patients experienced acute worsening of IPF (n=654, 30.7%). Additionally, 4.2% (n=90) of patients experienced more than three episodes of acute worsening. For initial acute worsening, idiopathic acute exacerbation was the most common cause (n=411, 19.2%), followed by acute exacerbation with pneumonia or non-pulmonary infection (n=112, 5.2%). Heart failure occurred in six patients. No episode of pulmonary embolism was observed.
Survival analysis was conducted only for patients diagnosed since 2016 because pirfenidone was under medical insurance coverage since October 2015. A total of 628 patients were diagnosed after January 1, 2016. They were followed up until data cutoff in October 2020. A total of 115 patients (18.3%) had died until data cutoff. The median survival time was 54 months since diagnosis. When assessing survival via Kaplan-Meier analysis, treatment with antifibrotic agents prolonged survival (p<0.001) (Figure 3). The median survival of the antifibrotics group was 54 months, longer than the median survival of 34 months of the non-antifibrotics group. Death was not observed in the no-treatment group. And the baseline lung functions of the no-treatment group were better than those of the treatment groups (Table 5).
The most common cause of death was idiopathic acute exacerbation (n=32), followed by acute exacerbation and pneumonia (n=18), pneumonia (n=13), and lung cancer (n=12). No patient died of pulmonary embolism. The reason for death was unknown for 22 patients.
Cox regression analysis was used to determine risk factors for mortality (Table 6). On univariate analysis, old age, walking distance and minimum oxygen saturation in six-minute walking test, lower initial FVC and DLco, lower GAP index, history of acute exacerbation, treatment without antifibrotics, and exposure to wood and stone dust were associated with higher mortality. However, only age, treatment without antifibrotics, history of acute exacerbation, and exposure to wood and stone dust were significantly associated with higher mortality on multivariate analysis.
Discussion
In this report, clinical characteristics, pattern of diagnosis, and treatment of Korean IPF patients in recent years were summarized. Compared with the previous Korean registry in 2008, the frequency of surgical lung biopsy has decreased and antifibrotics treatment has increased, especially since 2016. In addition, compared with registries from other countries, more patients were asymptomatic and at GAP stage 1 with higher DLco at the time of diagnosis. Based on these results, it can be assumed that Korean IPF patients tend to be diagnosed early.
It is well-known that IPF is more common in men and ever-smokers, with most of them being older than 50 years old. These characteristics were the same in this registry. The mean age of patients was 67.4±9.3 years, with a male preponderance (76.1%). The majority (71.0%) of patients were ever-smokers. All these findings were consistent with previous studies [7-10].
In this registry, a total of 1,755 patients reported about their occupations and dust exposure. In the previous Korean registry, some IPF patients reported that they had exposure to dust (13.0%). Dust-exposed occupations were associated with early onset of IPF and increased mortality [11]. About a quarter of patients had a history of exposures to wood, metal, fabric, or stone dust in this registry. As with the previous registry, exposure to wood and stone dust was also associated with increased mortality in the present registry. Compared with the previous Korean registry, fewer patients were engaged in primary industries, including agriculture and fishing, while more patients were exposed to dust. It might be because only university hospitals mainly located in large metropolitan areas have participated in the present registry.
The percentage of patients at GAP stage III (10.4%) was slightly increased than that in the previous registry (3.7%). However, compared with other registries from western countries, more patients were at GAP stage I. In this registry, 56.9% of patients were at stage I, whereas in a European registry, 20.4% and 29.7% of patients were at stage I and stage III, respectively [7]. In Korea, both previous and present registries included more GAP stage I patients than registries in other countries [12-14]. It might be due to early diagnosis of IPF in Korea. This might be because all Koreans have medical insurance and take many chest CTs during health examinations. Therefore, IPF could be diagnosed relatively early in Korea.
Early diagnosis can be assumed from pulmonary function tests and the proportion of initially asymptomatic patients, too. In this registry, the mean FVC was 74.6% and the DLco was 63.6%, similar to those of the previous Korean registry. However, they were slightly higher than previously published data from western countries, especially DLco. In earlier registries of Europe and North America, the mean DLco at diagnosis was 38.7%-57.3% [7,8,15,16]. Moreover, in this registry, 14.4% of patients were initially asymptomatic. Most asymptomatic patients might be diagnosed with IPF through health examinations.
The most common CT finding was honeycombing (67.0%). Additionally, most CT patterns were definite and probable UIP patterns. However, 5.8% of patients showed inconsistent UIP patterns. Although the most recently published international guideline [4] categorized HRCT findings into four patterns, at the time of planning this registry, a new guideline was not published. Therefore, in this registry, HRCT was classified using a previous guideline [5].
Changes of diagnostic method and treatment were noticeable between the previous registry and the present registry. About 20% of IPF patients were diagnosed after surgical lung biopsy in the current registry. Compared with the previous registry [17], the proportion of diagnosis with surgical lung biopsy was decreased. The decreasing frequency of surgical lung biopsy was also observed in other registries. Guenther et al. [8] reported that a surgical lung biopsy was performed in 32% in 2009, but in only 8% of cases in 2016. The authors thought it was possibly due to increased numbers of cryobiopsy [8]. However, in Korea, cryobiopsy was not perform popularly like that in Europe. The main reason in Korea might be the American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Latin American Thoracic Association guideline [5], which recommends not to conduct surgical lung biopsy for patients with a typical UIP pattern on HRCT.
Treatment was also markedly changed between current and previous registries in Korea. Treatment was mainly determined by the development of new medications and medical insurance coverage. The use of antifibrotics was increasing since 2012. In Korea, pirfenidone has been commercially available since late 2012. The medical insurance has started to cover pirfenidone since October 2015. Therefore, before 2012, the use of antifibrotic agents was very low. Since 2016, over 60% of patients were initially treated with antifibrotic agents.
Additionally, almost 80% of IPF patients were treated with an antifibrotic agent for over three months during the disease course. Considering that many patients discontinued pirfenidone due to side effects before three months, almost all IPF patients are thought to have tried antifibrotic treatment since 2016. Between the two antifibrotic agents, pirfenidone and nintedanib, pirfenidone has been used in most patients because of Korean medical insurance coverage. The declining usage of steroids, immunosuppressive agents, and N-acetylcysteine and the increase in the use of antifibrotic agents were reported in most other registries [8,18]. It reflects results of recent large randomized clinical trials and its subsequent subgroup analysis, real-world data of antifibrotics [2,3], the PANTHER-IPF trial [19], and changes in recent IPF guidelines.
Since 2016, 33 patients were simply followed up without treatment. It might be because they had a very early or minimal disease. In fact, the no-treatment group showed better pulmonary functions than the treatment groups at the time of initial diagnosis. In routine clinical practice, clinicians usually treat severe patients with medications and withholding treatment, including antifibrotic therapy for stable patients. A multicenter US registry reported that treatment at enrollment was associated with greater disease severity, more compromised quality of life, and the use of oxygen with activity [15]. No patient without treatment has died during the follow-up in this registry. It does not mean the “wait and see strategy” is beneficial for IPF patients. If patients become worse, the attending physician will start with the medication. Therefore, its real meaning is that some patients with IPF progress very slowly, although their chest CTs show a definite UIP pattern.
The survival of the antifibrotics group was longer than that of the non-antifibrotics group. It was already reported for this registry in a different time period [20]. This study proved the effectiveness of antifibrotics by analyzing patients diagnosed since 2016 when pirfenidone was started to be covered by medical insurance. Therefore, treatment bias due to economic problems was removed. The effect of antifibrotics on survival was also reported in other registries [7,8]. Behr et al. [7] reported that the risk of death was 37% lower in patients with antifibrotic therapy. Nintedanib and pirfenidone-treated patients were almost equally distributed in Behr et al.’s study [7]. Guenther et al. [8] reported that the median survival on antifibrotic drugs was 123.1 months compared with the median survival of 68.3 months in patients treated with any other medication, including immunosuppressive therapies. In the study of Guenther et al. [8], like the present registry, pirfenidone was used in the majority (83%) of patients.
Old age, history of acute exacerbation, and exposure to wood and stone dust were significantly associated with higher mortality. Additionally, treatment with antifibrotics was significantly associated with lower mortality. Several registries reported various but similar risk factors for mortality. In the previous Korean registry, age, FVC % predicted at diagnosis, and dust exposure were associated with mortality [11]. Behr et al. [7] reported that antifibrotic therapy, age, IPF disease duration, FVC% predicted, and overall physician’s judgment of rapid progression were assoicated with mortality. Jo et al. [18] reported that baseline lung function and GAP stage were strong predictors of mortality. Snyder et al. [14] reported that oxygen use at rest, lower FVC% predicted, and DLco% predicted were significantly associated with increased risk of death or lung transplantation. Regarding age, they reported an interesting result. The risk of death or lung transplantation increased with increasing age in patients ≥62 years old but decreased with increasing age in patients <62 years old [14]. The most interesting point about the risk factor analysis was that occupational dust exposure was associated with higher mortality in the current and previous Korean registries. Therefore, further study is needed for the relationship between occupational dust exposure and IPF.
This study has a few limitations. The major limitation of this study was that both existing and newly diagnosed patients were included, which might cause some bias for mortality. However, to minimize the “healthy survivor effect” and treatment bias due to medical insurance, only patients diagnosed since 2016 were included for survival analysis. Secondly, it was a retrospective study. Therefore, lots of follow-up data, for example, follow-up pulmonary function tests, were lost. Survival status was also potentially biased. However, this registry is being updated. Therefore, more accurate and long-term data will be revealed in the future. Furthermore, it will report more interesting and diverse analyses.
In conclusion, clinical characteristics of IPF patients in the present registry were similar to those in the previous Korean registry and registries from other countries. However, the frequency of surgical lung biopsy for diagnosis has decreased. Treatment with antifibrotics has also increased compared with the previous Korean registry. In addition, survival was significantly longer in the antifibrotics group than that in the non-antifibrotics group. Moreover, compared with registries of other countries, Korean IPF patients tended to be diagnosed early.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Data Sharing Statement
Data Sharing Statement Full text via DOI
Full text via DOI Print
Print Download Citation
Download Citation