Introduction
Bronchiectasis is defined as permanently dilated airways due to chronic bronchial inflammation caused by inappropriate clearance of various microorganisms and recurrent or chronic infection1. It was once considered to be an orphan disease with fading relevance in the developed world in the late 20th century2. Although the true prevalence is unknown for most regions, bronchiectasis is now being diagnosed with increasing frequency in North America and around the globe. The rate of bronchiectasis-associated hospitalizations increased markedly, starting around age 50, with the highest rate in the oldest age groups and particularly in older women in the United States3. Currently just under 1,000 people die from bronchiectasis each year, and the mortality rate also is increasing at 3% per year in England and Wales4. Clinical features of bronchiectasis include chronic production of sputum often mucopurulent or purulent in nature, persistent bacterial colonization and recurrent lower respiratory tract infections. Symptoms include hemoptysis and breathlessness characterized by mild to moderate airflow obstruction, lethargy and reduced health status5.
Bronchiectasis is a pathological description of lung damage, namely inflamed thick walled and dilated bronchi. Such damage is the ultimate consequence of inflammation and infection arising from various causes either inherited or acquired. Cystic fibrosis is the most common inherited cause of bronchiectasis in white populations, but there is increasing recognition of significant numbers of patients with bronchiectasis from a variety of causes6. Post-infectious damage is no longer the most common cause of bronchiectasis in developed countries following the introduction of antibiotic therapy and immunization programs. Irrespective of the underlying cause, patients with bronchiectasis are often colonized with bacterial species. This indicates failure of the usual host defense to maintain sterility of the respiratory tract, so the importance of identifying immunodeficiency has been underlined7.
In this review, we will focus on the immunological abnormalities that cause bronchiectasis in patients in whom cystic fibrosis has been excluded, and discuss several controversies in the current management of this disorder.
Treatable Causes of Bronchiectasis
The underlying conditions associated with bronchiectasis are listed in Table 1. It is an important question whether knowledge of the underlying medical cause leads to a change in management of patients with bronchiectasis. One study conducted in two pediatric respiratory clinics identified the etiology in 77% of cases with non-cystic fibrosis bronchiectasis. Furthermore, they found that immunodeficiency and intrinsic abnormalities accounted for the majority of cases and that knowledge of a specific causal agent led to a modification in management8. This has been confirmed in the adult setting in a study performed at a tertiary hospital in the United Kingdom9. Among 165 patients confirmed to have bronchiectasis on computed tomography scan, an underlying cause was identified in 74% of patients. Knowledge of the underlying cause directly affected management in 37% of patients. These studies suggest that investigation of the underlying causes of bronchiectasis leads to an alteration in therapy to target these specific conditions in many more cases than was previously thought, which can have significant prognostic implications. As the treatable causes of bronchiectasis, primary immune deficiency, allergic bronchopulmonary aspergillosis, nontuberculous mycobacterial infection, airway obstruction, inflammatory bowel disease, rheumatoid arthritis and aspiration can be included10.
Immune Deficiencies in Non-Cystic Bronchiectasis
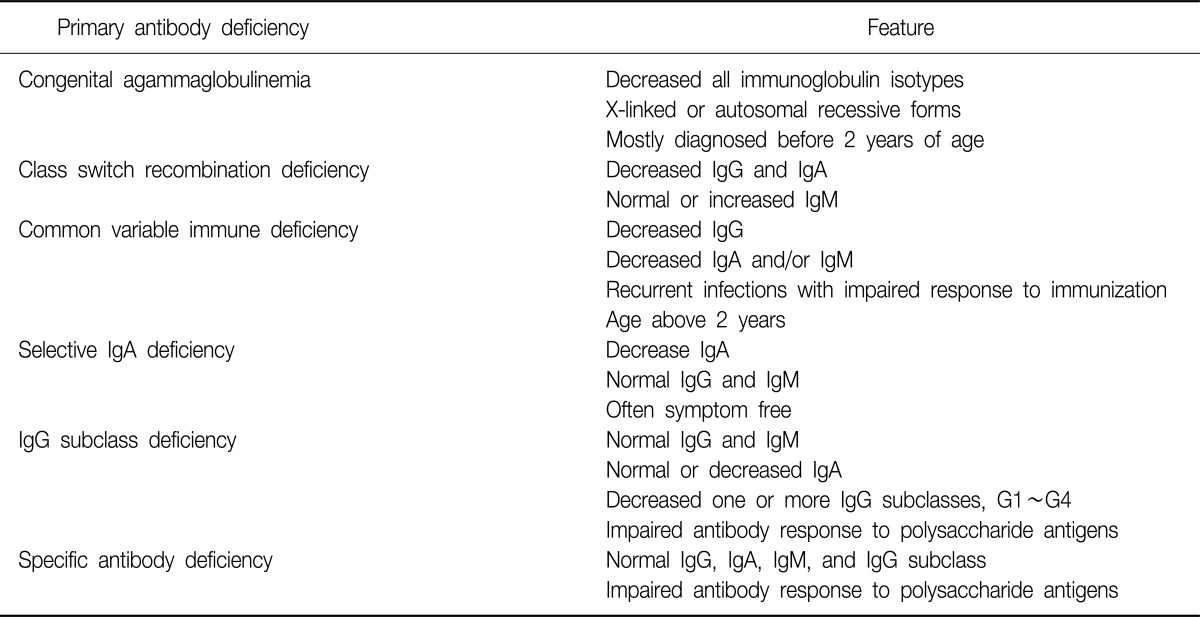
A range of immunologic abnormalities are associated with non-cystic fibrosis bronchiectasis, and the prevalence of each in bronchiectasis varies from study to study11. Primary antibody deficiency is the most common primary immunodeficiencies12. It is characterized by a defect in the production of normal amounts of antigen-specific antibodies. These antibodies or immunoglobulins are essential for the adaptive immune response against various pathogens. A defect in antibody production causes recurrent and severe infections. Primary antibody deficiency represents a heterogeneous spectrum of conditions, including congenital agammaglobulinemia, common variable immunodeficiency, class switch recombination deficiency, selective immunoglobulin A deficiency, specific antibody deficiency, and immunoglobulin G subclass deficiency13. The key features of primary antibody deficiencies are summarized in Table 2.
Congenital agammaglobulinemia is a genetic defect which impairs antibody production of all immunoglobulin isotypes and response to vaccinations. X-linked agammaglobulinemia accounts for 85% of all cases of congenital agammaglobulinemia, and autosomal recessive forms account for the rest12. Class switch recombination deficiencies, formerly known as hyper-immunoglobulin M syndromes, are very rare conditions characterized by decreased serum immunoglobulin G and A levels, but increased immunoglobulin M levels14. Common variable immunodeficiency is an idiopathic antibody deficiency, defined by serum immunoglobulin G levels below 2 SD of normal controls in the presence of decreased immunoglobulin A and/or immunoglobulin M levels, recurrent infections, impaired response to immunization, exclusion of defined causes of hypogammaglobulinemia, and an age above 2 years15. These 3 entities (congenital agammaglobulinemia, class switch recombination deficiency, and common variable immunodeficiency) have been commonly associated with development of bronchiectasis as a clinical complication12.
Selective immunoglobulin A deficiency is defined as a decrease of serum immunoglobulin A levels below 2 SD of age-matched controls (or less than 0.07 g/L) but normal serum immunoglobulin G and M levels12,16. The clinical course is asymptomatic in many patients, and only a minority of patients develops recurrent lower respiratory tract infections and/or bronchiectasis12.
Immunoglobulin G subclass deficiency is defined as an abnormally low level of one or more immunoglobulin G subclasses in patients with normal levels of total immunoglobulin G and M. Immunoglobulin A level may also be low. Some patients with subclass deficiency exhibit impaired specific antibody production16. By the way, there is some controversy over whether deficiencies of immunoglobulin G subclasses have a role in the development of bronchiectasis. In a study estimating immune function in adults with bronchectasis, a number of subjects (14%) had low level of immunoglobulin G3, but the significance of this finding was uncertain17. Another small, single center, case-control study showed that almost half (48%) of the patients with bronchiectasis of unknown etiology have low serum concentrations of one or more immunoglobulin G subclasses, and the patients with immunoglobulin subclass deficiency revealed impaired antibody response to H. influenza type b conjugated vaccine18. A study recently conducted in Korea also showed that the frequency of immunoglobulin G subclass deficiency was high (45%) in patients with bronchiectasis of unclear etiology19. These results suggests that immunoglobulin G subclass deficiency is not an unusual cause of bronchiectasis, and that detailed investigation of humoral immune status, including the level of immunoglobulin G subclasses and antibody response to specific antigen is needed in cases of bronchiectasis without definite causes.
Finally, specific antibody deficiency is characterized by normal concentrations of immunoglobulin G, A, M and immunoglobulin G subclasses and abnormal antibody responses to polysaccharide vaccines16. The prevalence is unknown, but it may be a frequent finding in patients evaluated for recurrent respiratory tract infections. It has been proven through a number of studies that specific antibody deficiency is a recognized cause of bronchiectasis9,20,21.
The interpretation of anti-pneumococcal antibody concentration results is based on antibody increases over pre-immunization concentrations and on final concentrations following immunization. Traditionally, adequate responses to individual pneumococcal serotypes were defined as a post-immunization antibody concentration of 1.3 g/mL or higher or at least 4-fold over baseline22, but recent two reports have shown new cutoffs for identifying individual with an inadequate response to vaccine23,24.
Treatment of Non-Cystic Fibrosis Bronchiectasis
Bronchiectasis occurring unrelated to cystic fibrosis is a common and difficult respiratory condition to manage. However, it has historically received little attention, and many of the recommendations for its management have been extrapolated from the studies for the management of cystic fibrosis rather than based on appropriate research25. In 2010, British Thoracic Society extensively reviewed this condition and first developed a comprehensive guideline for its management26. In this section, we will discuss recent evidences of therapeutic options available to treat patients with non-cystic fibrosis bronchiectasis.
1. Intravenous immunoglobulin therapy
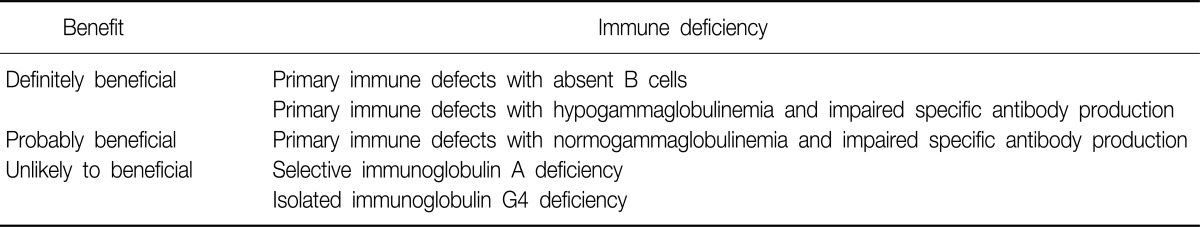
Intravenous immunoglobulin is indicated as replacement therapy for patients with primary and selected secondary immunodeficiency diseases characterized by absent or deficient antibody production with recurrent or severe infections27. The uses of intravenous immunoglobulin in primary immune deficiencies were summarized in Table 3. The role of immunoglobulin therapy is clear in patients with common variable immunodeficiency or agammaglobulinemia aiming to reduce the frequency of infectious episode and to prevent further destruction of the airway27-29. Immunoglobulin replacement therapy also should be provided in normogammaglobulinemic patients with polysaccharide nonresponsiveness and evidence of recurrent infections16,27. However, selective immunoglobulin A deficiency is not an indication for immunoglobulin replacement therapy27, and the cautious use is recommended in selected patients with isolated immunoglobulin subclass deficiency16.
In a recent study, response to intravenous immunoglobulin therapy was estimated in adult patients with recurrent infections and isolated immunoglobulin G3 subclass deficiency30. As we have described above, this condition can be a cause of bronchiectasis in adults. The result was that the majority of patients showed significant clinical improvement with a decrease in frequency and severity of infections. This study offers the potential of intravenous immunoglobulin as a treatment option in patients with immunoglobulin subclass deficiency and recurrent infections.
2. Airway pharmacotherapy
1) Bronchodilators
Although a large proportion of subjects with bronchiectasis have airflow obstruction with airway hyperreactivity and a significant bronchodilator response, there are no randomized, controlled trials investigating the effects of long-acting beta-agonists or anticholinergics in the management of patients with bronchiectasis31,32.
Tiotropium bromide, a long-acting muscarinic receptor antagonist, relaxes airway smooth muscle cells and suppresses airway submucosal gland secretions. Nowadays, it is widely used as one of the important medications in patients with chronic obstructive pulmonary disease, but the efficacy in bronchiectasis has not been studied adequately. In a small open label Japanese study, tiotropium improved symptoms of cough, sputum, and dyspnea in patients with chronic mucus hypersecretion, but too small numbers of patients with bronchiectasis were included33. A recent trial in Chinese showed that one month of inhalation of tiotropium improved the clinical symptoms and body mass index, airflow obstruction, dyspnea, and exercise capacity (BODE) index of the patients with bronchiectasis34. Randomized, controlled trials will be needed to evaluate the effectiveness of long-acting beta-agonists and anticholinergics in the treatment of bronchiectasis.
2) Inhaled corticosteroids
Inhaled corticosteroids have proved undeniable benefits in patients with asthma or chronic obstructive pulmonary disease, but very little is known of its anti-inflammatory effects in bronchiectasis. Several small scale studies have observed that in patients with non-cystic fibrosis bronchiectasis, high doses of inhaled corticosteroids can positively influence several bronchial inflammatory parameters and certain key symptoms, such as dyspnea or sputum volume, while improving patients' health-related quality of life, without affecting either the number of exacerbations or lung function35-37. However, a Cochrane review concluded that there was insufficient evidence to recommend the routine use of inhaled corticosteroids in adults with stable-state bronchiectasis, and that a therapeutic trial may be justified in adults with difficult to control symptoms and in a certain subgroups38.
Meanwhile, the addition of a long-acting beta-agonist to inhaled corticosteroids proved to reduce the dose of inhaled corticosteroids without effects of treatment in chronic obstructive pulmonary disease or asthma39. In actual practices, it is prescribed to a high percentage of patients with bronchiectasis without any clear scientific evidence. A recent, small-sized, 12-month randomized trial revealed that inhaled medium-dose formoterol-budesonide combined treatment is more effective in symptom control and health-related quality of life compared with high-dose budesonide treatment in patients with non-cystic fibrosis bronchiectasis40. Largerscale studies of longer duration are needed to confirm this result.
3) Inhaled antibiotics
There are a substantial number of literatures on the use of prolonged antibiotics in patients with bronchiectasis, because reducing the bacterial burden in the airways may decrease inflammation and promote healing of the bronchial tree41. These studies showed small benefit in response rates and sputum scores, but did not show any differences in exacerbation rates, lung function, or quality of life scores42.
Similarly, there are also a large number of publications concerning the use of inhaled antibiotics (mainly tobramycin and gentamicin) in patients with bronchiectasis, particularly in the setting of Pseudomonas aeruginosa infection43,44. Some benefits have been documented in these studies, including a decrease in bacterial density, airway inflammation, and exacerbation frequency. However, the benefits appear to be less than in cystic fibrosis cases, and bronchospasm appear to be more common in adults with non-cystic fibrosis bronchiectasis than reported in the cystic fibrosis population45, and treatment needs to be continuous for its ongoing efficacy44. More recently, new inhaled agents such as liposomal ciprofloxacin and liposomal amikacin are also being investigated for potential use in bronchiectasis.
3. Macrolides as an anti-inflammatory therapy
The mechanism of the beneficial effect of macrolide antibiotics in patients with bronchiectasis and other chronic inflammatory airway diseases is most likely related with their immunomodulatory properties in addition to the antimicrobial activities46. The trials of macrolide treatment in bronchiectasis are limited in number, size of study population, and length of treatment and follow up. Although the effect of macrolide therapy on lung function remain uncertain, there is consistent evidence of a decrease in inflammation, frequency of exacerbation, and sputum volume47. These findings need to be confirmed in larger studies with longer follow-up times and careful assessment of harmful effects in order to define a role for macrolides in the treatment of bronchiectasis.
Caution also needs to be exercised if the possibility of nontuberculous mycobacterial infection exists in a patient for whom macrolide therapy is being contemplated because macrolide monotherapy increases the likelihood of emergence of macrolide resistance, which would be extremely problematic to treat48,49. Newer macrolides are being developed that have only immunomodulatory properties50.
4. Mucolytics
Mucus retention in the lungs is a prominent feature of bronchiectasis. The stagnant mucus becomes chronically colonized with bacteria, which elicit a host neutrophilic response. This fails to eliminate the bacteria, and the large concentration of host-derived protease may contribute to the airway damage.
Mucolytic drugs have anti-inflammatory, antioxidant and mucoregulatory properties. Some benefits in exacerbation rate and lung function have been demonstrated in chronic obstructive pulmonary disease51. Among the mucolytics, erdosteine is a thiol derivative agent approved for use in chronic obstructive pulmonary disease in some European countries. A small randomized study suggests that erdosteine decreases cough, dyspnea, and sputum in elderly patients with bronchiectasis and chronic mucus hypersecretion52.
Inhalation of osmotic agents, such as hypertonic saline and mannitol is known to accelerate tracheobronchial clearance in many conditions, probably by increasing the osmolarity of the airway surface fluid and creating a driving force for water to move quickly into the airway lumen53,54. Based on the available evidence, mannitol dry powder inhalation is well tolerated and improves the quality of life of patients with non-cystic fibrosis bronchiectasis55. There is a need for well designed and adequately powered multicentre trials to establish the potential usefulness of mannitol.
5. Chest physiotherapy
Effective clearance of mucus from the airways may break the vicious cycle of the disease process by decreasing the stagnation of mucus and mucus plug formation with associated bacterial colonization, recurrent infection, and inflammation.
Chest physiotherapy has been used for many years and a number of techniques are available for mobilizing secretions, such as postural drainage, active cycle of breathing techniques, positive expiratory pressure, oscillatory positive expiratory pressure devices and high-frequency chest wall percussion56. One recent, small, randomized control trial concluded that regular chest physiotherapy using an oscillatory positive expiratory pressure device in patients with non-cystic fibrosis bronchiectasis had small, but significant benefits in improving cough severity, exercise capacity, and SGRQ total score despite earlier negative systematic reviews57,58.
6. Vaccination
There are no randomized controlled trials examining the effectiveness of influenza vaccination in patients with bronchiectasis, so no evidences for or against routine influenza vaccination59. There is limited evidence to support the use of 23-valent pneumococcal vaccination as routine management in adults and children with bronchiectasis, because a Cochrane review found only one single randomized trial that fulfilled the inclusion criteria60. It was open label and examined 167 patients with bronchiectasis61. Over a 2-year period, the study found a significant reduction in acute infective exacerbations in the group immunized with the 23-valent pneumococcal vaccine and influenza vaccine compared with the group receiving the influenza vaccine alone. Therefore, it can be suggested that influenza vaccination should also be administered in patients with bronchiectasis.
In addition, patients with bronchiectasis associated with selective antibody deficiency may benefit from additional immunization with conjugate pneumococcal vaccines16. Patients who fail to respond to the polysaccharide vaccine when immunized after 2 years of age usually respond to the conjugate vaccine62.
Conclusion
Awareness of non-cystic fibrosis bronchiectasis has increased over the past few years with a concomitant increase in publications. Investigation of the underlying cause of bronchiectasis is the most important key to effective management. The uses of intravenous immunoglobulin is helpful in reducing the frequency of infection and preventing further airway destruction in selected patients with primary immune deficiencies. The effectiveness of long-acting beta agonists with or without inhaled corticosteroid and long-acting anticholinergics is uncertain. New inhaled antibiotics under investigation are expected to be used for controlling recurrent infections. Patients with selective antibody deficiency may benefit from additional immunization with conjugate pneumococcal vaccines. There is a clear need for well designed large-scale randomized trials to answer these questions regarding the optimum therapeutic approach for the patients with bronchiectasis.

 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Print
Print Download Citation
Download Citation