Introduction
Chronic obstructive pulmonary disease (COPD) is associated with persistent respiratory symptoms and airflow limitations due to damage to the airways and alveolar structures caused by exposure to noxious particles or gases [1]. The prevalence, morbidity, and mortality from COPD are very high; thus, COPD represents an important socioeconomic and public health burden [2]. Patients with COPD have several comorbidities, such as coronary artery disease, diabetes, osteoporosis, sarcopenia, and depression, and the ailment is recognized as a systemic disease due to systemic inflammation [3,4].
Various mechanisms explain the causes of systemic inflammation, the most common of which is oxidative stress [5]. Reactive oxygen species (ROS) form through in vivo metabolic processes that use oxygen as a fuel, as well as during exposure to external or toxic environments [6]. An optimal level of ROS is beneficial for maintaining biological activities by inducing inflammation, immune function, autophagy, and a proper stress response [7]. However, over-activation of ROS leads to DNA damage and cell apoptosis and causes ischemic injury at the tissue level [8].
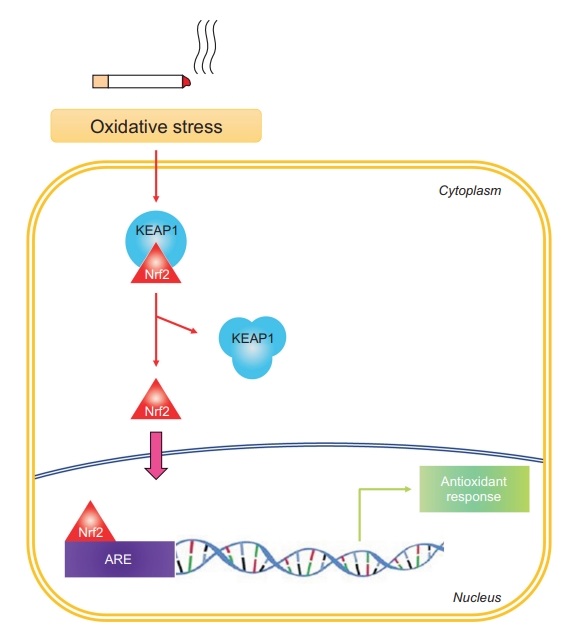
Smoking is the main cause of COPD, as it generates excessive oxidative stress, causing alveolar epithelial cell damage and triggering the development and progression of COPD [9]. Several systems in the body fight this excessive oxidative stress. The most representative is the nuclear factor erythroid 2‒related factor 2 (Nrf2) pathway, which belongs to the cap ‘n’ collar subfamily of basic region leucine zipper transcription factors. Nrf2 induces several drug-metabolizing enzymes, such as glutathione S-transferase and NAD(P) H:quinone oxidoreductase 1 by upregulating a common DNA sequence called the antioxidant response element (ARE) [10]. Nrf2 is degraded proteasomally by its cytosolic inhibitor Kelch-like ECH-associated protein 1 (KEAP1) under basal conditions and is released from KEAP1 under oxidative stress conditions to activate the ARE (Figure 1) [11].
Nrf2 is a representative antioxidant molecule that has been emphasized for its relevance to COPD and significant association with oxidative stress. Furthermore, research has been actively conducted on the possibility that Nrf2 is a COPD biomarker and on its role as a therapeutic target. In this paper, we review the significance of oxidative stress and Nrf2 in the development and progression of COPD and summarize the value of Nrf2 as a COPD biomarker.
Nrf2 in the Development of COPD
Long-term exposure of a normal airway to smoking has a fundamental impact on the development of COPD, leading to a chronic inflammatory reaction, resulting from deposition of various inflammatory cells. However, destruction of lung structure and resulting emphysema or small airway obstruction does not occur in all smokers [12,13]. Many hypotheses have been proposed about the causes of COPD in some smokers, including mucus hyper-production due to abnormal immune reactions, small airway obstruction due to excessive tissue repair and airway remodeling; insufficient tissue repair leads to emphysematous changes [14].
Especially, many animal studies have been conducted on the role of Nrf2 in the development of emphysema, a major phenotype of COPD. A large number of researchers insist that inadequate tissue repair due to loss of the protective role of Nrf2 leads to emphysema-dominant COPD. Nrf2-knockout mice is a useful tool to evaluate the role of Nrf2. Nrf2-knockout mice with a BALB/c background can be established by specific deletion of the Nrf2 gene segment. Nrf2-knockout mice grow normally and are fertile, but are susceptible to oxidative stress. Nrf2-knockout COPD model was made by intratracheal instillation of elastase or exposure to cigarette smoke (CS) in Nrf2-knockout mice. Iizuka et al. [15] and Ishii et al. [16] showed that neutrophilic inflammation and elastase activity increase in bronchoalveloar lavage (BAL) fluid after exposing Nrf2-knockout mice to CS, which reduces antioxidant and anti-protease activities. These results suggest that functional loss of Nrf2 is involved in COPD development in smokers [15,16]. In another study conducted by Sussan et al. [17], destruction of alveolar structure and pulmonary hypertension are attenuated in wild-type Nrf2 mice exposed to CS after oral administration of the triterpenoid CDDO-imidazolide Nrf2 activator.
The role of Nrf2 in the development of COPD has also been studied in humans. Goven et al. [18] compared the expression of Nrf2 and Keap1 in lung tissues and alveolar macrophages of patients with and without emphysema. They showed that Nrf2 protein expression decreased, whereas the expression of Keap1 increased. They suggested that this altered equilibrium of Nrf2-Keap1 and reduction in the expression of sequential antioxidant proteins was the main mechanism of emphysematous changes in smokers [18]. Suzuki et al. [19] compared Nrf2 expression in alveolar macrophages obtained from BAL fluid among young subjects, older current smokers, and patients with COPD. They found that Nrf2 expression decreased in the macrophages of older current smokers and patients with COPD [19].
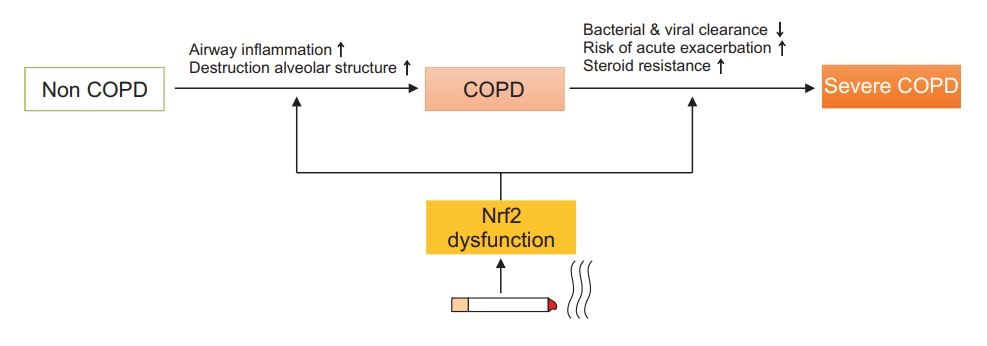
Most of these studies suggest that a lack of tissue repair and chronic inflammation via downregulation of Nrf2 lead to obstruction of small airways and destruction of lung parenchyma, resulting in the development of COPD.
Nrf2 in the Progression of COPD
The most important factor leading to disease progression during the natural course of COPD is an exacerbation. Frequent exacerbations of COPD affect not only acute symptoms but also reduce long-term lung function [20]. Furthermore, an exacerbation increases the morbidity and mortality from COPD [21]. COPD exacerbations are triggered mainly by acute airway inflammation caused by bacterial or viral infection [22]. Most patients with COPD are susceptible to bacterial or viral infections. Studies have shown that oxidative stress resulting from chronic exposure to smoke or noxious particles leads to dysfunction of the alveolar macrophages, which impairs phagocytic activity [23]. Therefore, downregulation of Nrf2 in patients with COPD may lead to excessive oxidative stress, dysfunction of alveolar macrophages, COPD exacerbation, and disease progression. Several studies have experimentally demonstrated this hypothesis. Harvey et al. [24] showed that pulmonary bacterial clearance of alveolar macrophages by phagocytosis in Nrf2 wild-type and Nrf2-deficient mice improves by activating Nrf2 with the isothiocyanate sulforaphane, suggesting that Nrf2 could be a major target of exacerbation in relation to bacterial clearance. Degradation of the host defense due to reduced Nrf2 has also been studied in viral infections. Cho et al. [25] showed that clearance of respiratory syncytial virus (RSV) decreases and excessive virus-induced inflammation is induced in Nrf2-deficient mice. Recurrent RSV infection in the COPD mouse model induces inflammatory cytokine production, such as interleukin (IL)-1α, IL-17, and interferon-γ, as well as the expression of proteases, such as matrix metalloproteinase-2 and -8, resulting in destruction of lung tissues [26].
Thus, these studies suggest that a decrease in Nrf2 activity may be a major factor in the progression of COPD (Figure 2). They also suggest that activation of Nrf2 expression may inhibit the progression of COPD. Based on these concepts, Wise et al. [27] conducted a randomized, double-blind, placebo-controlled trial to determine whether oxidative stress, airway inflammation, and pulmonary function improve with the use of the Nrf2 inducer oral sulforaphane in 89 patients with COPD. However, the effects of sulforaphane in patients with COPD were not demonstrated. It was also unclear whether activation of Nrf2 was ineffective for COPD patients because sulforaphane failed to stimulate the expression of Nrf2 target genes or some other problem. Further clinical trials of COPD using another Nrf2 activator are needed. To date, there has been significant research on the role of Nrf2 in inhibiting COPD progression in vivo, but negative data have been reported for clinical implications. Therefore, further studies are needed to clarify the role of Nrf2 in COPD progression.
Nrf2 and the Severity of COPD
As stated in the Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines, patients with COPD are treated according to disease severity and classified as to their airflow limitation, risk of exacerbation, and symptom burden. However, since the 2017 update, the degree of airflow limitation represented by forced expiratory volume in 1 second (FEV1) in the GOLD guidelines has been excluded from the patient classification criteria. However, FEV1 remains a good indicator of an individual patient’s clinical outcome [28]. Many studies have classified severity of COPD according to the extent of these airflow limitations, and have investigated whether differences in oxidative stress, proteostasis imbalance, and structural changes in the lung are among them. Min et al. [29] showed that Nrf2 and histone deacetylase 2 (HDAC2) expression decrease with increasing severity of COPD, according to airflow limitation (GOLD I-IV) in paraffin-embedded lung sections of patients with COPD. They also documented increased oxidative stress and corticosteroid resistance along with COPD severity.
Exacerbation of COPD has been defined as worsening of the respiratory symptom beyond normal day-to-day variations. Many studies have revealed that COPD exacerbation is closely related to excessive inflammation in response to increased oxidative stress. It is thus important to understand the mechanism of exacerbation and role of Nrf2 during the acute exacerbation. Frequent exacerbations are closely related to the severity of COPD, and long-term macrolide treatment, such as azithromycin, lowers the incidence of exacerbations. Therefore, in the GOLD 2019 guidelines, azithromycin is recommended for use with maximal inhaler treatment for frequent exacerbators [30]. The phosphoinositide 3-kinase (PI3K)/Akt pathway is upregulated by excessive oxidative stress in patients with COPD, which reduces the expression of HDAC2. The use of a macrolide inhibits inflammation by inhibiting the PI3K/Akt pathway and restoring corticosteroid sensitivity by upregulating HDAC2 [31,32]. Nrf2 acts as a key factor downstream of PI3K/Akt. However, the relationship between the PI3K/Akt-Nrf2 pathway and airway and systemic inflammation has not yet been investigated well in severe COPD patients. Therefore, further studies are required to detail the mechanisms of the Nrf2 and PI3K/Akt pathways in patients with severe COPD.
Nrf2 as a Diagnostic and Treatment Target in Patients with COPD
Some studies have predicted the clinical outcomes, disease severity, and exacerbation risk in patients with COPD using Nrf2, and some have assessed the possible relationships between polymorphisms in the Nrf2 gene and COPD. Yamamoto et al. [33] compared the polymorphisms of the Nrf2 promoter region in 87 patients with COPD and 50 healthy controls, but they did not find any statistical difference among them. However, in a study of 915 subjects on the general population of Japan, a specific single nucleotide polymorphism of the Nrf2 gene (rs6726395) was associated with a decline in FEV1, particularly in the major allele of smokers [34]. The researchers insisted that smoking affects the Nrf2 polymorphism, which leads to a decrease in FEV1 and ultimately to the development of COPD.
Several studies have reported increased oxidative stress due to impaired Nrf2 expression in the bronchial epithelium and lung tissue of patients with COPD. Yamada et al. [35] reported that Nrf2 mRNA and protein expression significantly decrease in bronchial epithelial cells of COPD patients. As previously mentioned, Nrf2 expression decreases according to the degree of airway limitation in the lung tissues of patients with COPD [29]. The attenuation of Nrf2 at the airway and tissue levels causes the development and progression of COPD.
The concept of COPD as a systemic inflammatory disease of oxidative stress, rather than a simple airway inflammatory disease, has implications for peripheral blood Nrf2 levels. Fratta Pasini et al. [36] showed that Nrf2 expression increases in peripheral blood mononuclear cells of patients with COPD, suggesting that this is a systemic response to oxidative stress that persists even after the cessation of smoking. Indeed, plasma Nrf2 levels are negatively correlated with FEV1 and FEV1/forced vital capacity but increase during exacerbation in COPD patients [37]. The findings of these two studies contrast with the general trend in Nrf2 expression described above. However, the Nrf2 expression pattern may differ between the airway and systemic inflammatory process in COPD patients, so further study is needed to resolve the precise mechanism.
Conclusion
The key molecule controlling oxidative stress from smoking or noxious particles is Nrf2. Genetic changes and dysfunction of Nrf2 induce chronic inflammation, apoptosis, and proteolysis of alveolar cells, resulting in changes in the airway and lungs. Furthermore, Nrf2 accelerates disease severity and the progression of COPD (Figure 3). Nrf2 has been emphasized as a potential biomarker for predicting the initiation, severity, and degree of airway or systemic inflammation in COPD, even as a treatment target to improve airway limitations or prevent deterioration of COPD. However, COPD is not a simple airway disease but rather a systemic disease, and Nrf2 is more complex when responding to local and systemic oxidative stress. Therefore, additional studies to uncover the precise mechanisms of Nrf2 are needed to use Nrf2 as a diagnostic and therapeutic biomarker for COPD in clinical practice.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Print
Print Download Citation
Download Citation